Biologická terapie systémového lupus erythematodes – zaměřeno na belimumab
Belimumab je prvním biologickým lékem schváleným v USA, Kanadě a Evropě k terapii systémového lupus erythematodes (SLE) a prvním novým léčivem v této indikaci po více než padesáti letech. Jde o plně humánní rekombinantní IgG1λ monoklonální protilátku blokující vazbu solubilního BLyS (stimulátor B-lymfocytů, soluble B lymphocyte stimulator) na B-lymfocyty. V současnosti je belimumab schválen Evropskou lékovou agenturou (EMA) v indikaci přídatné léčby dospělých pacientů se SLE s pozitivními autoprotilátkami s vysokým stupněm aktivity choroby bez přítomnosti aktivního postižení ledvin či centrálního nervového systému. Aktuálně probíhají další studie i u pacientů s aktivní lupusovou nefritidou a u dalších autoimunitních onemocnění (Sjögrenův syndrom, ANCA asociované vaskulitidy) a dalších stavů.
Úvod
Systémový lupus erythematodes (SLE) je autoimunitní systémové onemocnění často se projevující postižením více orgánových systémů (kůže, klouby, kardiovaskulární systém, ledviny, centrální i periferní nervový systém, plíce a další), které je doprovázeno výraznými abnormalitami imunitního systému. Jde o vzácné onemocnění, které postihuje častěji ženy; poměr dospělých žen a mužů se SLE je 9 : 1 [1]. Patogenetických mechanismů vzniku SLE je studováno mnoho, jedním z nich je porucha interakce mezi T-lymfocyty a B-lymfocyty ve smyslu mj. defektní apoptózy, přežívání autoreaktivních klonů, nadprodukce autoprotilátek, poruchy signálních molekul a jejich receptorů. B-lymfocyty představují základní buňky protilátkové imunity, podílejí se na tvorbě autoprotilátek a regulaci imunitní odpovědi. Prostřednictvím prezentace antigenu a kostimulačních molekul indukují aktivaci a diferenciaci T-lymfocytů.
Zcela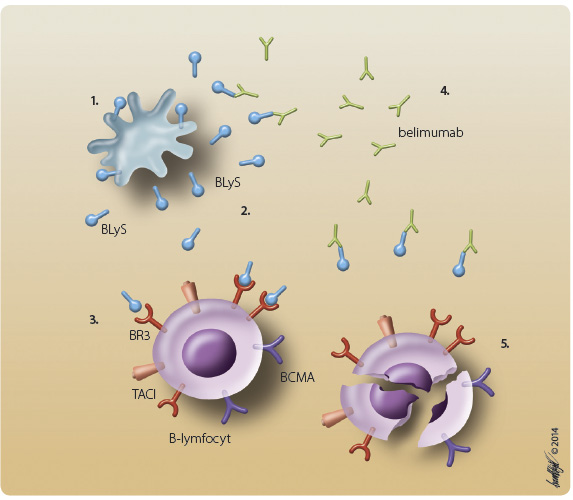 novým terapeutickým přístupem v terapii SLE je blokáda vazby solubilního BLyS (stimulátor B-lymfocytů, soluble B lymphocyte stimulator), označovaného rovněž zkratkou BAFF (B-lymfocyty aktivující faktor, B-cell activating factor), na B-lymfocyty (obr. 1) [2]. BLyS patří mezi cytokiny řazené do rodiny TNF (tumor necrosis factor) a je exprimován na povrchu buněk B-buněčné linie, působí jako jejich silný aktivátor. Hraje také zásadní roli při B-buněčné proliferaci a diferenciaci [3].
novým terapeutickým přístupem v terapii SLE je blokáda vazby solubilního BLyS (stimulátor B-lymfocytů, soluble B lymphocyte stimulator), označovaného rovněž zkratkou BAFF (B-lymfocyty aktivující faktor, B-cell activating factor), na B-lymfocyty (obr. 1) [2]. BLyS patří mezi cytokiny řazené do rodiny TNF (tumor necrosis factor) a je exprimován na povrchu buněk B-buněčné linie, působí jako jejich silný aktivátor. Hraje také zásadní roli při B-buněčné proliferaci a diferenciaci [3].
U pacientů se SLE byly popsány jeho zvýšené hladiny spolu s akcentovanou buněčnou expresí. Přestože však byl prokázán vztah mezi hladinami BLyS a aktivitou SLE, není dosud role BLyS v patogenezi SLE zcela jasná [4].BLyS je za účasti dalších zánětlivých cytokinů (IL-2, IFNγ, TNFα) štěpen na solubilní proteinovou část. Na povrchu B-lymfocytů je BLyS vázán třemi buněčnými receptory označovanými jako BLyS receptor 3 (BR3), transmembránový aktivátor s modulátorem vápníku a cyklofilin ligand interaktor (TACI, transmembrane activactor and calcium modulator and cyclophilin ligand interactor) a antigen maturace B-lymfocytů (BCMA, B-cell maturation antigen), z nichž nejsilnější vazbu má receptor BR3 [5, 6]. Blokáda této vazby vede k apoptóze a inhibici maturace B-lymfocytů. Po podání belimumabu dochází k selektivní supresi CD20 pozitivních B-lymfocytů a plazmatických buněk s krátkým poločasem [2, 5].
Farmakokinetika a farmakodynamika
Belimumab je rekombinantní lidský imunoglobulin IgG1λ o molekulární hmotnosti 147 kDa, který vytváří komplexy se solubilní formou BLyS a inhibicí její vazby na buněčné receptory vede k supresi, snížení diferenciace, aktivace a přežívání B-lymfocytů. Belimumab se neváže na B-lymfocyty přímo, ale prostřednictvím vazby na BLyS. U pacientů se SLE léčených belimumabem byly v klinických studiích zjištěny výrazně snížené hladiny cirkulujících C19+, CD20+ aktivovaných B-lymfocytů [2, 7]. Při dlouhodobém klinickém sledování byly statisticky významně snížené hladiny B-lymfocytů v 52. týdnu léčby (p = 0,008). Studie prokázaly, že belimumab ovlivňuje nejdříve pokles počtu nově aktivovaných B-lymfocytů (ve 12. až 14. týdnu, p < 0,05) a až poté pokles počtu plazmatických a paměťových buněk (v 76. týdnu; p < 0,01 a p < 0,05) [7].
Belimumab v intravenózní dávce 10 mg/kg dosahoval maximálních sérových koncentrací (rozmezí: 173–573 μg/ml) krátce po ukončení aplikace. Sérová koncentrace belimumabu klesala bioexponenciálně s distribučním poločasem 42 hodin a s terminálním poločasem 19,4 dne. Clearance se pohybovala v rozmezí 69–622 ml/den. Celkový distribuční objem belimumabu činil 5,29 litru. Přesná cesta eliminace belimumabu není plně objasněna [8, 9].
Bezpečnost a interakce
Bezpečnost belimumabu se neliší od bezpečnosti biologických přípravků ovlivňujících B-lymfocyty. Klinické studie fáze II a III přinesly při sledování nežádoucích účinků belimumabu poměrně příznivé výsledky [2, 7].
U celkem 2133 pacientů se SLE sledovaných ve studiích fáze II a III byl hlášen výskyt nežádoucích účinků jakékoli závažnosti. Nežádoucí příhody byly zaznamenány u 93 % pacientů ze skupiny, která dostávala placebo, a u 92 % pacientů ze skupiny léčené belimumabem. Nejčastěji byly hlášeny: nevolnost (15 %), průjem (12 %), pyretická reakce (10 %), nazofaryngitida (9 %), bronchitida (9 %), nespavost (7 %), deprese (5 %), bolesti hlavy (5 %), cystitida (4 %), leukopenie (4 %) a virová gastroenteritida (3 %). V průběhu klinických sledování hodnotících účinnost a bezpečnost belimumabu u SLE bylo zaznamenáno celkem 10 případů malignit, z toho 5 nádorů solidních orgánů (dva nádory prsu, jeden nádor žaludku, jeden nádor děložního čípku a jeden nádor ovaria) a 5 nemelanomových nádorů kůže. Hematologické malignity ve sledované skupině zaznamenány nebyly. V průběhu sledování bylo dokumentováno celkem 15 úmrtí. Tři úmrtí byla zaznamenána v placebové skupině ve studii BLISS-52 v důsledku infekčních komplikací a kardiovaskulární příhody, jedno úmrtí nastalo z neobjasněné příčiny. Ve skupině pacientů léčených belimumabem v dávce 1 mg/kg bylo celkem šest úmrtí (maligní tumor, iktus, suicidium, infekce, komplikace základní choroby, z neobjasněné příčiny) a ve skupině léčené dávkou 10 mg belimumabu/kg bylo také šest úmrtí (komplikace choroby, celkem třikrát infekční komplikace, suicidium, kardiovaskulární komplikace) [7, 10, 11].
V klinických studiích u pacientů se SLE nebyly prokázány interakce či změny farmakokinetických vlastností chronicky podávaných léčiv (antimalarika, glukokortikoidy, azathioprin, methotrexát, mykofenolát mofetil, inhibitory ACE, nesteroidní antiflogistika a statiny). Výrobce nedoporučuje kombinaci belimumabu s dalšími léky biologické povahy, jejichž cílem jsou B-lymfocyty (anti-CD20 – rituximab) kvůli zvýšení nárůstu rizika závažných infekcí. Ze stejného důvodu není doporučována ani konkomitantní terapie cyklofosfamidem. Nevhodná je vakcinace živými vakcínami při probíhající léčbě belimumabem, vzhledem k typu probíhající imunosuprese [7, 8, 11].
Indikace a dávkování
Americký úřad FDA (U.S. Food and Drug Administration) schválil 9. března 2011 belimumab k terapii SLE. V návaznosti na toto schválení proběhla úspěšná registrace i v České republice, v jednání je dosud úhrada belimumabu. Česká revmatologická společnost (ČRS) vypracovala indikační kritéria podávání belimumabu vycházející z doporučení FDA [12]. Podávání belimumabu není zatím indikováno a není obecně doporučováno u pacientů se SLE s těmito orgánovými projevy: aktivní neurolupus s postižením centrálního nervového systému, aktivní lupusová nefritida III., IV. a V. typu vyžadující razantní imunosupresi dle protokolu, přítomnost proteinurie s hodnotou vyšší než 6 g/24 h a elevace hladin kreatininu nad 220 μmol/l.
Lupusová nefritida per se však nepředstavuje kontraindikaci léčby belimumabem, kontraindikací je jen aktivní lupusová nefritida. Pacient s lupusovou nefritidou s normální renální funkcí a non-nefrotickou proteinurií může být pro extrarenální indikace belimumabem léčen. U pacientů, kteří trpí hypogamaglobulinemií s hodnotami imunoglobulinu G nižšími než 400 mg/dl (IgG < 400 mg/dl) nebo deficiencí IgA (IgA < 10 mg/dl), se belimumab nedoporučuje podávat. Vylučujícím kritériem pro terapii je přítomnost hepatitidy B a C, pozitivita HIV či závažná probíhající infekce. Rovněž jsou vyloučeni pacienti se známou alergií na složky podávaného léčiva [13].Belimumab je indikován u pacientů se SLE s klinickou i humorální aktivitou bez přítomnosti vylučovacích kritérií. Musí být přítomna pozitivita antinukleárních protilátek (ANA)
a/nebo anti-dsDNA protilátek a snížení hladin komplementu. Aktivita choroby hodnocená skórovacím systémem SELENA-SLEDAI (SELENA, Safety of Estrogens in Lupus Erythematosus National Assessment; SLEDAI, Systemic Lupus Erythematosus Disease Activity Index) musí mít hodnotu vyjádřenou jako 6 nebo více. Pokud po šesti měsících léčby belimumabem nedojde ke klinickému ani laboratornímu zlepšení hodnocenému pomocí skóre SELENA-
-SLEDAI ≥ 4bodové snížení SELENA-SLEDAI skóre nebo dojde k nárůstu skóre, jsou tito pacienti považováni za non-respondéry a terapie belimumabem by měla být ukončena [8, 11–13].
Terapie belimumabem je u pacientů se SLE zahajována indukční dávkou 10 mg/kg podávanou ve dnech 0, 14 a 28, která je následována udržovací terapií, kdy je infuze podávána ve 4týdenních intervalech. Před podáním infuze belimumabu se doporučuje podat antihistaminikum s antipyretikem k minimalizaci poinfuzních reakcí [13].
Klinické studie
Bezpečnost a účinnost léčby belimumabem byla prokázána v klinických studiích fáze I a II a ve dvou studiích fáze III trvajících 52 a 76 týdnů. Nyní jsou k dispozici i data ze sedmiletého sledování hodnotícího dlouhodobou účinnost a bezpečnostní profil belimumabu.
Klinické studie fáze I
Primárním cílem randomizované, multicentrické a dvojitě slepé studie fáze I (LBSL01) bylo stanovení bezpečnosti a snášenlivosti léčby belimumabem pacienty se SLE. Do studie bylo zařazeno celkem 70 pacientů se SLE splňujících podmínky určené studijním protokolem (stabilní klinická aktivita a léčba SLE, absence podávání cyklofosfamidu, nepřítomnost aktivní lupusové nefritidy a postižení centrálního nervového systému, nepřítomnost závažných infekcí). Pacienti byli randomizováni do skupin léčených belimumabem (57 pacientů) a placebové skupiny (13 pacientů). Belimumab byl podáván v dávkách 1 mg/kg, 4 mg/kg, 10 mg/kg a 20 mg/kg v jedné nebo dvou aplikacích ve dni 0 a následně 21. den. Léčba byla dobře tolerována a incidence nežádoucích příhod se proti placebové skupině nelišila. Byl dokumentován statisticky signifikantně významný pokles počtu CD20+ B-lymfocytů u pacientů po jedné dávce belimumabu proti placebu (den 42, p = 0,0042; den 84, p = 0,0036), následně po dvou dávkách (den 105, p = 0,0305). Ovlivnění aktivity SLE belimumabem nebylo prokázáno, nicméně data získaná touto studií poskytla cenné údaje pro další fáze klinického zkoušení II a III [2, 7].
Studie fáze II
Randomizovaná, multicentrická a dvojitě slepá studie fáze II (LBL02) vyhodnocovala snášenlivost a účinnost belimumabu u pacientů s aktivním a středně aktivním SLE bez přítomnosti závažných orgánových manifestací (ledviny, centrální nervový systém). Ve studii bylo celkem 449 pacientů randomizovaně rozděleno do skupin dle podávané dávky belimumabu (1 mg/kg, 4 mg/kg a 10 mg/kg) a do placebové skupiny. K hodnocení odpovědi na terapii byl použit skórovací systém SELENA-SLEDAI a skupina pacientů léčených belimumabem vykazovala 19,5% zlepšení proti původním hodnotám. Medián doby do vypuknutí prvního vzplanutí nemoci (tzv. flare) byl 83 dní u pacientů léčených belimumabem, ale 67 dní ve skupině pacientů dostávajících placebo. Pacienti léčení belimumabem měli statisticky signifikantně nižší výskyt vypuknutí flare v průběhu 24. až 52. týdne (154 dní vs. 108 dní, p = 0,0361).
Pokles počtu CD20+ společně s poklesem hladiny anti-dsDNA protilátek byl statisticky signifikantní ve skupině s belimumabem proti placebu v týdnu 52. Mezi skupinami pacientů se výskyt nežádoucích příhod nelišil.
Studie LBL02 nedosáhla svého primárního cíle, nebyla prokázána výrazně vyšší efektivita belimumabu proti placebu. Předpokládanou příčinou bylo malé omezení konkomitantní terapie a nevhodné nastavení primárních cílů, které mělo rozlišovat více mezi skupinou léčenou belimumabem a placebovou skupinou. Nicméně výsledky LBL02 umožnily plynulý přechod ke studiím fáze III [14].
Studie fáze III
BLISS-52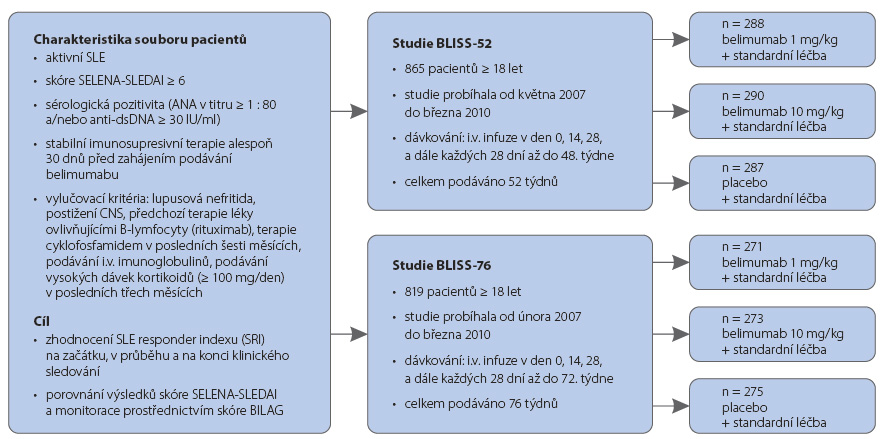 a BLISS-76 (obr. 2) byly studie fáze III určené pro dospělé pacienty s aktivním SLE s protilátkovou aktivitou se stabilní imunosupresivní terapií (skóre ≥ 6 dle SELENA-SLEDAI, se sérologickou pozitivitou antinukleárních protilátek – ANA – v titru ≥ 1 : 80 a s pozitivitou anti-dsDNA protilátek ≥ 30 IU/ml). Do studie nebyli zařazeni pacienti se závažnými orgánovými komplikacemi (aktivní lupusová nefritida, postižení CNS) a s předchozí terapií léky ovlivňujícími B-lymfocyty (rituximab) či s terapií cyklofosfamidem v posledních šesti měsících, pacienti léčení intravenózními imunoglobuliny nebo vysokými dávkami steroidů (> 100 mg/den) v posledních třech měsících. Obě studie fáze III porovnávaly účinnost a bezpečnost belimumabu podávaného intravenózně v dávkách 1 mg/kg a 10 mg/kg, a to ve dnech 0, 14 a 28, dále pak každých 28 dní (podávání ve studii BLISS-52 bylo ukončeno ve 48. týdnu, ve studii BLISS-76 v 72. týdnu) ve srovnání s placebem.
a BLISS-76 (obr. 2) byly studie fáze III určené pro dospělé pacienty s aktivním SLE s protilátkovou aktivitou se stabilní imunosupresivní terapií (skóre ≥ 6 dle SELENA-SLEDAI, se sérologickou pozitivitou antinukleárních protilátek – ANA – v titru ≥ 1 : 80 a s pozitivitou anti-dsDNA protilátek ≥ 30 IU/ml). Do studie nebyli zařazeni pacienti se závažnými orgánovými komplikacemi (aktivní lupusová nefritida, postižení CNS) a s předchozí terapií léky ovlivňujícími B-lymfocyty (rituximab) či s terapií cyklofosfamidem v posledních šesti měsících, pacienti léčení intravenózními imunoglobuliny nebo vysokými dávkami steroidů (> 100 mg/den) v posledních třech měsících. Obě studie fáze III porovnávaly účinnost a bezpečnost belimumabu podávaného intravenózně v dávkách 1 mg/kg a 10 mg/kg, a to ve dnech 0, 14 a 28, dále pak každých 28 dní (podávání ve studii BLISS-52 bylo ukončeno ve 48. týdnu, ve studii BLISS-76 v 72. týdnu) ve srovnání s placebem.
Primárním cílem obou studií bylo zhodnocení tzv. SLE responder indexu (SRI) na začátku, v průběhu a na konci klinického sledování, dále porovnání výsledků skóre SELENA-SLEDAI a monitorace prostřednictvím skóre BILAG (British Isles Lupus Assessment Group), zaměřeného na výskyt nových či zhoršení stávajících manifestací SLE [15, 16].
Studie BLISS-52
Studie BLISS-52 zahrnovala celkem 865 pacientů rozdělených do skupin podle dávky belimumabu (1 mg/kg, n = 288 pacientů; 10 mg/kg, n = 290 pacientů) a do placebové skupiny (n = 287 pacientů). Rozdíl v počtu pacientů, kteří ukončili studii kvůli neefektivitě belimumabu oproti placebu, nebyl statisticky signifikantní (4,17 % při podávání dávky 1 mg/kg, 4,14 % při podávání dávky 10 mg/kg vs. 5,57 % při podávání placeba). Terapeutická odpověď hodnocená indexem SRI v týdnech 28 a 52 byla statisticky signifikantně vyšší ve skupinách léčených belimumabem než v placebové skupině (p < 0,05). Rovněž u skupiny pacientů léčených belimumabem bylo pomocí skórovacího indexu SELENA-SLEDAI zaznamenáno zlepšení hodnocené jako pokles o více než 4 body v týdnu 52 proti placebu. Flare se objevil u 70 % pacientů ve skupině léčené belimumabem v dávce 1 mg/kg (p = 0,0026) a u 71 % pacientů ve skupině léčené dávkou 10 mg/kg (p = 0,0036) proti 80% výskytu u pacientů ve skupině s placebem. Průměrná doba do výskytu prvního flare byla při léčbě belimumabem 126 dní (dávka 1 mg/kg) a 119 dní (dávka 10 mg/kg), zatímco ve skupině placeba byla průměrná doba jen 84 dní. V laboratorním hodnocení byl zaznamenán statisticky významný pokles hladiny anti-dsDNA protilátek až jejich vymizení u pacientů léčených belimumabem proti placebu (p < 0,02). Byla zjištěna normalizace hladin komplementu 4 (C4) při terapii belimumabem proti placebu (p < 0,03) a statisticky signifikantní zvýšení hladin C3 bylo zjištěno jen ve skupině pacientů léčených dávkou 10 mg/kg proti placebu (p = 0,0005). Dobrá terapeutická odpověď ve skupině s belimumabem umožnila částečnou detrakci steroidní terapie. Belimumab se zdál účinnější než placebo při zhodnocení skórovacích indexů aktivity a laboratorních parametrů v 52. týdnu léčby. Rozdíly v účinnosti dle dávky belimumabu nebyly statisticky signifikantní. Výskyt nežádoucích příhod byl obdobný ve skupinách léčených belimumabem v dávkách 1 mg/kg a 10 mg/kg a v placebové skupině. Byly pozorovány závažné infekce v jednotlivých skupinách u 22, 13 a 17 pacientů. Hypersenzitivní reakce na podání infuze byly stejné v obou skupinách s belimumabem, v každé z nich u dvou pacientů [15, 16].
Studie BLISS-76
Studie BLISS-76 zahrnovala celkem 819 pacientů rozdělených do skupin podle podávané dávky belimumabu (1 mg/kg, n = 271 pacientů; 10 mg/kg, n = 273 pacientů) a do placebové skupiny (n = 275 pacientů). Rozdíl v počtu pacientů, kteří ukončili studii z důvodu neefektivity belimumabu vůči placebu, nebyl statisticky signifikantní (4,43 % při podávání dávky 1 mg/kg, 6,23 % při dávkování 10 mg/kg belimumabu vs. 7,27 % při podávání placeba). Terapeutická odpověď hodnocená indexem SRI v týdnu 52 a 76 byla vyšší ve skupinách léčených belimumabem než v placebové skupině, nicméně rozdíl nebyl statisticky signifikantně významný. Skupina pacientů léčených belimumabem prokázala i pomocí skórovacího indexu SELENA-SLEDAI zlepšení hodnocené jako pokles o více než 4 body v týdnu 52 proti placebu, ale výsledek nebyl statisticky signifikantní. Při hodnocení v 76. týdnu byl nalezen statisticky signifikantní rozdíl mezi skupinou dostávající belimumab v dávce 1 mg/kg a placebovou skupinou (42,1 % vs. 33,8 %, p < 0,05). Skupina pacientů dostávajících dávku 10 mg/kg nevykazovala statisticky signifikantní rozdíl proti placebu (41,4 %, p = nesignifikantní). Odpověď hodnocená skórovacím systém BILAG u pacientů léčených dávkou 1 mg/kg belimumabu byla v 52. týdnu 74,9 %, zatímco při podávání placeba 65,5 % (p < 0,05) a v 76. týdnu činil tento poměr 69,0 % vs. 58,9 % (p < 0,05). Zlepšení hodnocené skórovacím systémem SF-36 PSC (36-item Short Form Physical Component Score), které bylo zaznamenáno ve skupině s dávkou 1 mg/kg (+4,37) oproti skupině s placebem (+2,85) v týdnu 52, bylo statisticky významné (p = 0,012). Ve 28. a 76. týdnu nebyl zaznamenán statisticky signifikantní rozdíl mezi těmito skupinami ani mezi skupinou s dávkou 10 mg/kg a skupinou s placebem. Výskyt závažných vzplanutí nemoci byl ve skupinách léčených belimumabem statisticky signifikantně nižší proti placebové skupině. V laboratorním hodnocení byl zaznamenán statisticky významný pokles hladiny anti-dsDNA protilátek až jejich vymizení u pacientů v terapii belimumabem (1 mg/kg) proti placebu v týdnech 52 a 76 (p < 0,01), respektive tomu tak bylo i při podávání dávky 10 mg/kg (p < 0,001). Byla zaznamenána normalizace hladin komplementu 4 (C4) při terapii belimumabem proti placebu (p < 0,03). U hladin C3 bylo pozorováno zvýšení hodnocené v týdnu 52 a 76 ve skupině s dávkou 1 mg/kg proti placebu, nicméně bez statistické významnosti, naopak ve skupině s dávkou 10 mg/kg bylo zvýšení oproti placebu statisticky významné, a to 43,5 % (p < 0,01), respektive 51,3 % (p < 0,001). Dobrá terapeutická odpověď ve skupině s belimumabem umožnila částečné snížení dávek steroidní terapie. Belimumab se zdál účinnější než placebo při zhodnocení skórovacích indexů aktivity a laboratorních parametrů v 52. a 76. týdnu léčby. Rozdíly v účinnosti dle dávky belimumabu nebyly statisticky signifikantní. Výskyt nežádoucích příhod byl obdobný ve skupinách léčených belimumabem v dávkách 1 mg/kg a 10 mg/kg a v placebové skupině [15, 16].
Post hoc analýza studií BLISS-52 a BLISS-76
Velmi zajímavá data týkající se změn renálních parametrů přinesla post hoc analýza výsledků studií BLISS-52 a BLISS-76. Obě dvě studie nebyly primárně zaměřeny na posouzení vlivu belimumabu na projevy lupusové nefritidy, aktivní lupusová nefritida byla i mezi vylučujícími kritérii pro zařazení do klinického hodnocení. Pacienti léčení belimumabem měli příznivější výsledky renálních parametrů (proteinurie, glomerulární filtrace, renální relapsy, sérologická aktivita choroby) než nemocní v placebové skupině, ačkoli rozdíly mezi oběma skupinami nebyly statisticky signifikantní. U pacientů s renálním postižením přítomným již při vstupu do studie, kteří byli léčeni mykofenolátem, došlo k výraznějšímu zlepšení renálních parametrů, pokud současně užívali belimumab. Výsledky této analýzy naznačují, že terapie belimumabem by mohla být pro pacienty s renálním postižením přínosem, limitací je však malý počet pacientů, a je proto třeba dalších hodnocení [17].
Použití v graviditě a laktaci
O použití belimumabu v těhotenství je k dispozici zatím jen malé množství údajů. Studie na zvířatech u opic neukázaly na přímé ani nepřímé škodlivé účinky belimumabu s ohledem na reprodukční toxicitu, belimumab je dle klasifikace FDA hodnocen v kategorii C. V platném SPC se uvádí, že ženy ve fertilním věku musí během léčby a po dobu nejméně 4 měsíců od ukončení léčby užívat účinnou antikoncepci [13]. Přesto došlo k desítkám gravidit, jejichž výsledky zatím nejsou příliš povzbudivé [14]. To však nelze v současnosti připisovat nežádoucím účinkům belimumabu, ale spíše nepříznivému vlivu základního onemocnění [18]. V těhotenském registru výrobce je v současnosti zahrnuto šest pacientek a zatím bylo referováno o narození dvou zdravých dětí [19]. O užití belimumabu v době kojení je ještě méně údajů. Dle SPC se doporučuje při rozhodnutí o kojení zvážit prospěch z kojení pro dítě a prospěch z léčby pro ženu [13]. Byl prokázán přestup belimumabu do mateřského mléka opic, není ale známo, zda se po požití systémově vstřebává. Vzhledem k velikosti molekuly se však dá očekávat jen minimální přestup belimumabu do mateřského mléka a jeho degradace v zažívacím traktu kojence.
Závěr
Belimumab představuje inovativní léčivo indikované k terapii SLE. Jde o humánní monoklonální protilátku, která inhibuje biologickou aktivitu stimulátoru B-lymfocytů (BLyS). Belimumab je v současnosti indikován k terapii aktivního SLE, nejsou-li přítomny vylučující orgánové komplikace (aktivní lupusová nefritida – s deteriorací renálních funkcí a nefrotickou proteinurií, neurolupus). Pacient s lupusovou nefritidou, normální renální funkcí a non-nefrotickou proteinurií může být pro extrarenální indikace belimumabem léčen. Terapie belimumabem byla ve více studiích účinná, dobře tolerovaná a bezpečná. V současnosti belimumab není lékem první volby, ale je užíván v kombinaci s dalšími imunosupresivy (např. s azathioprinem, mykofenolátem) k udržení remise.Zatím nejsou k dispozici relevantní data o účinnosti a bezpečnosti belimumabu u pacientů s aktivní lupusovou nefritidou a postižením CNS, zde je třeba dalších hodnocení s větším počtem pacientů.V České republice je belimumab již registrován, není však zatím stanovena jeho úhrada, a vedle klinických studií je proto podáván jen na základě schválení revizním lékařem zdravotní pojišťovny. Obě pracoviště autorů tohoto sdělení již mají s podáváním belimumabu zkušenosti. Širšímu využití této léčby zatím brání nejen absence stanovené úhrady, ale také skutečnost, že belimumab je určen pro skupinu pacientů, kteří mají již jiné terapeutické alternativy, a dále chybění přesvědčivých dat u těžkých forem SLE se závažnými orgánovými komplikacemi.
Belimumab je dále zkoušen a zdá se perspektivní v některých dalších indikacích, a to nejen revmatologických (Sjögrenův syndrom, ANCA asociované vaskulitidy), ale i transplantologických a neurologických (např. myasthenia gravis).
Seznam použité literatury
- [1] Urowitz M, Gladman D, Ibanez D, et al. Evolution of disease burden over 5 years in a multicentre inception SLE cohort. Arthritis Care Res (Hoboken) 2011.
- [2] Baker KP, Edwards BM, Main SH, et al. Generation and characterization of LymphoStat-B, a human monoclonal antibody that antagonizes the bioactivities of B lymphocyte stimulator. Arthritis Rheum 2003; 48: 3253–3265.
- [3] Treml JF, Hao Y, Stadanlick JE, et al. The BLyS family: toward a molecular understanding of B cell homeostasis. Cell Biochem Biophys 2009; 53: 1–16.
- [4] Jakob N, Stohl W. Cytokine disturbances in systemic lupus erythematosus. Arthritis Res Ther 2011; 13: 228.
- [5] Cancro MP. The BLyS/BAFF family of ligands and receptors: key targets in the therapy and understanding of autoimmunity. Ann Rheum Dis 2006; 65 (Suppl 3): 34–36.
- [6] Elsawa SF, Novak AJ, Grote DM, et al. B-lymphocyte stimulator (BLyS) stimulates immunoglobulin production and malignant B-cell growth in Waldenstrom macroglobulinemia. Blood 2006; 107: 2882–2888.
- [7] Furie R, Stohl W, Ginzler EM, et al. Belimumab Study Group. Biologic activity and safety of belimumab, a neutralizing anti-B-lymphocyte stimulator (BLyS) monoclonal antibody: a phase I trial in patients with systemic lupus erythematosus. Arthritis Res Ther 2008; 10: 109.
- [8] Lamore R, Parmar S, Patel K, Hilas O. Belimumab (Benlysta): a breakthrough therapy for systemic lupus erythematosus. P T 2012 ;37: 212–226.
- [9] Halpern WG, Lappin P, Zanardi T, et al. Chronic administration of belimumab, a BLyS antagonist, decreases tissue and peripheral blood B-lymphocyte populations in cynomolgus monkeys: pharmacokinetic, pharmacodynamic, and toxicologic effects. Toxicol Sci 2006; 91: 586–599.
- [10] Jacobi AM, Huang W, Wang T, et al. Effect of long-term belimumab treatment on B cells in systemic lupus erythematosus: extension of a phase II, double-blind, placebo-controlled, dose-ranging study. Arthritis Rheum 2010; 62: 201–210.
- [11] Merrill JT, Ginzler EM, Wallace DJ. Long-term safety profile of belimumab plus standard therapy in patients with systemic lupus erythematosus. Arthritis Rheum 2012 Jun 5. doi: 10.1002/art.34564.
- [12] Horák P, Tegzová D, Závada J, et al. Doporučení ČRS pro léčbu nemocných se SLE. Čes Revmatol 2013; 21: 110–122.
- [13] European Medicines Agency. Benlysta (belimumab): EU Summary of Product Characteristics. http://www.ema.europa.eu/docs/cs_CZ/document_library/EPAR_-_Product_Information/human/002015/WC500110150.pdf Navštíveno 30. května 2014.
- [14] Wallace DJ, Stohl W, Furie RA, et al. A phase II, randomized, double-blind, placebo-controlled, dose-ranging study of belimumab in patients with active systemic lupus erythematosus. Arthritis Rheum 2009; 61: 1168–1178.
- [15] Navarra SV, Guzmán RM, Gallacher AE, et al. BLISS-52 Study Group. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet 2011; 377: 721–731.
- [16] Hahn BH. Belimumab for Systemic Lupus Erythematosus. N Engl J Med 2013; 368: 1528–1535.
- [17] Furie R, Petri M, Zamani O, et al. A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. BLISS-76 Study Group. Arthritis Rheum 2011; 63: 3918–3930.
- [18] Peart E, Clowse ME. Systemic lupus erythematosus and pregnancy outcomes: an update and review of the literature. Curr Opin Rheumatol 2014; 26:118–123.
- [19] Landy H, Powell M, Hill D, et al. Belimumab pregnancy registry: prospective cohort study of pregnancy outcomes. Obstet Gynecol 2014; 123 Suppl 1: 62S.