Faktory ovlivňující biologickou dostupnost léčiv
Souhrn:
Biologická dostupnost je jedním z farmakokinetických ukazatelů hodnotících množství léčiva, které se dostane v aktivní formě do systémového oběhu. Je ovlivněna řadou faktorů, a to jak na straně léčiva či lékové formy, tak na straně organismu. Výsledný efekt léčiva u konkrétního pacienta může být velmi odlišný od teoretického předpokladu, zejména ovlivněním jeho absorpce.
Key words: bioavailability – absorption – solubility – pH.
Summary:
Bioavailability is one of the pharmacokinetic parameters used in the evaluation of the amount of drug reaching systemic circulation in active form. It's affected by a number of factors related both to drug or its form and organism itself. The resulting drug’s effect in a particular patient can differ substantially from the theoretical presumption, mainly due to modification of their absorption.
Farmakokinetické metody se snaží matematickými vztahy vystihnout časovou závislost pobytu léčiva v organismu [1]. Vycházejí z analýzy koncentrací léčiva a jeho metabolitů v dostupných tělních tekutinách (nejčastěji krev, moč a sliny). Mezi ně řadíme informace o biologické dostupnosti, o distribuci léčiva v organismu a o rychlosti jeho eliminace [1]. Využíváme je k predikci koncentrace léčiva a k individualizaci dávkování. Hovoříme o koncentračním profilu léčiva, který je popsán pomocí základních farmakokinetických parametrů. Ty závisejí na řadě fyziologických i patofyziologických procesů v organismu a na faktorech na straně léčiva. Parametry rozdělujeme na primární a sekundární [1]. Mezi primární ukazatele řadíme distribuční objem (Vd) a clearance (Cl), jejichž změny lze přímo vysvětlit změnami fyziologických proměnných (jako např. průtokem krve, vazbou na bílkoviny, glomerulární filtrací). Sekundární parametry, mezi něž patří biologická dostupnost (F), biologický poločas eliminace (t1/2), plocha pod křivkou plazmatické koncentrace (AUC), závisejí na parametrech primárních [1].
Biologická dostupnost
Množství léčiva z dávky obsažené v podaném léčivém přípravku, které se dostává ve farmakologicky aktivní (ve většině případů metabolicky nezměněné) formě do systémového krevního oběhu, se nazývá biologická dostupnost (F) [1]. Vyjadřuje se relativně, tj. jako část z dávky, a může tedy nabývat hodnot od 0 do 1 (nebo rovněž od 0 do 100 %) a zahrnuje v sobě aspekt kvantitativní (míru biologické dostupnosti) a rychlostní. Rozlišujeme absolutní a relativní biologickou dostupnost [1].
Absolutní biologická dostupnost je definována jako absolutní část z podané dávky, která se z lékové formy (po jiném než intravenózním podání) dostane do systémového krevního oběhu. Absolutní biologická dostupnost po intravenózním podání je rovna hodnotě 1, resp. 100 %. Úbytek aktivní formy léčiva před dosažením systémového oběhu je cennou informací i z ekonomického hlediska (nízká biologická dostupnost zvyšuje náklady na léčbu).
Relativní biologická dostupnost představuje relativní část dávky léčiva, která se dostává do systémového krevního oběhu při srovnání testované lékové formy s jinou lékovou formou (obě jsou jiné než pro intravenózní podání) [1].
Biologická dostupnost je ovlivněna farmaceutickou a farmakokinetickou fází cesty léčiva organismem [1]. Farmaceutická fáze rozhoduje o farmaceutické dostupnosti, tj. o množství léčiva uvolněného z léčivého přípravku a o rychlosti, kterou se tento děj uskutečňuje. Rozhoduje o množství léčiva, které je dostupné pro průnik biomembránou. Zde je třeba zmínit farmaceutické děje dezintegrace, dezagregace a disoluce, které biologickou dostupnost mohou značně ovlivnit [1]. Farmakokinetická fáze pak zahrnuje absorpci, distribuci a eliminaci (biotransformaci a exkreci). Mezi faktory ovlivňující absorpci léčiv po perorálním podání můžeme zařadit fyzikálně chemické vlastnosti samotného léčiva ovlivňující transport přes membrány, změny v rozpustnosti léčiva v místě absorpce, typ lékové formy, příjem a druh potravy, biorytmy a intra a interindividuální rozdíly v lidské populaci.
Faktory ovlivňující biologickou dostupnost
Fyzikálně chemické vlastnosti léčiva
Mezi fyzikálně chemickými vlastnostmi léčiva jmenujme rozpustnost ve vodě a v tucích, acidobazické vlastnosti (pH a pKa), rozdělovací koeficient, molekulovou hmotnost, tvar molekuly a míru schopnosti vázat se na plazmatické proteiny [2]. Obecně lze říci, že léčivo je těžce vstřebatelné, pokud je jeho molekulová hmotnost > 500, dekadický logaritmus rozdělovacího koeficientu oktanol/voda > 5 a molekula má 5 a více vodíkových donorů [2].
Biofarmaceutický klasifikační systém
(BCS) rozděluje léčivé látky do čtyř tříd na základě
jejich rozpustnosti ve vodném prostředí
a gastrointestinální propustnosti (permeability) [2,3],
tabulka 1. Za vysoce
rozpustná jsou považována ta léčiva, jejichž nejvyšší
dávka se rozpustí v 250 ml vodného pufru v rozmezí
pH 1–8, za vysoce propustná jsou označována ta léčiva,
jejichž absorpce z gastrointestinálního traktu (GIT)
u člověka dosahuje nejméně 90 % z podané dávky
[2].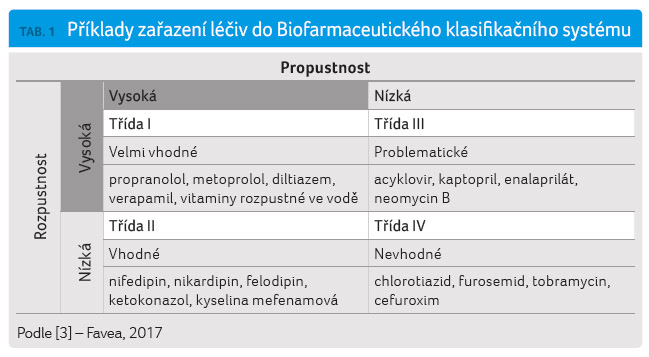
Rozpustnost v tucích
Rozpustnost v tucích určuje schopnost léčiva difundovat přes lipofilní membrány [2]. Míra absorpce pasivním transportem, respektive prostou difuzí, klesá se snižující se rozpustností látky v lipidech. Lépe se absorbují látky v nedisociované formě, struktura určuje, zda a v jakém prostředí zůstane léčivo natolik apolární (rozpustné v tucích), aby mohlo procházet lipofilními buněčnými membránami. Tato vlastnost se uplatňuje např. u inhalačních anestetik, jejichž účinek stoupá se vzrůstající lipofilitou [4]. Lipofilitu a hydrofilitu léčiva charakterizuje disociační konstanta (pKa), tj. hodnoty pH, při němž se 50 % léčiva nachází v ionizované (hydrofilní) podobě [2,5].
Acidobazické vlastnosti léčiva
Je li pH prostředí rovno disociační konstantě léčiva (pKa), je 50 % léčiva ve formě ionizované a 50 % ve formě neionizované. U látky kyselé povahy platí, že čím je pH vyšší (prostředí zásaditější), tím vyšší je míra ionizace; naopak u zásad platí, že čím je pH nižší (prostředí kyselejší), tím je disociace zásad větší. Ionizovaná forma léčiva je hydrosolubilní a lipoidními membránami neprostupuje [2,5].
Rozdělovací koeficient
Rozdělovací koeficient (log P) mezi vodnou a nepolární fází (nejčastěji n oktanol) je důležitý pro rychlost prostupu léčiv přes biologické membrány [2,5]. Čím je log P vyšší, tím je látka lipofilnější a snadněji proniká do tkání.
Velikost a tvar molekuly
Většina terapeuticky používaných léčiv má molekulovou hmotnost pohybující se v rozmezí 100–1 000 daltonů (Da) [5]. Některá antibiotika mají z tohoto důvodu nulovou biologickou dostupnost, nevstřebávají se vůbec a musejí být do systémového řečiště podávána přímo (i.v.), aby dosáhla systémového účinku, nelze je tedy podávat v jiné lékové formě, např. perorální. Uveďme např. vankomycin, molekulová hmotnost je vyšší (1 450 Da), perorální cestou se nevstřebává, jeho biologická dostupnost je nulová, perorální podání není tedy spojeno s rizikem orgánové toxicity (pouze v ojedinělých případech toto riziko může existovat). A právě tato nulová biologická dostupnost se využívá u léčby průjmů způsobených Clostridium difficile. Podává se prášek pro injekci (v dávce 500 mg), který se rozpustí ve vodě a pacient ho vypije s odstupem vždy 6 hodin. Vankomycin tak působí pouze lokálně.
Větších molekulových hmotností dosahují například peptidy; menší peptidy mívají molekulovou hmotnost kolem 1 000 Da, větší i přes 10 000 Da [5]. Jako příklad uveďme biosimilární přípravky zaváděné do biologické léčby. Jedná se o léčiva s tak velkou molekulovou hmotností, že převyšují molekulovou hmotnost chemických léčiv řádově o desítky až stovky Da, jejich struktura je velmi složitá [6]. Primární struktura proteinové molekuly léčiva je dána sekvencí bází, ale sekundární a terciární a posttranslační modifikace jsou závislé na produkující buněčné linii a na podmínkách, ve kterých je udržována [6]. Z toho vyplývají možné odlišnosti obou skupin – originálních biotechnologických léčiv a biosimilars – v imunogenním profilu, terapeutickém efektu i v riziku vzniku nových nežádoucích účinků [6]. Výjimečně jsou do organismu podávána léčiva, jejichž molekulová hmotnost je ještě vyšší (trombolytické enzymy kolem 50 000 Da) [5].
Vazba na plazmatické bílkoviny
Lipofilní látky mají vyšší tendenci vázat se na plazmatické bílkoviny než látky hydrofilní [5]. Vazba vzniká rychle a je reverzibilní. Jedná se o depotní systém, který na jedné straně snižuje intenzitu účinku léčiva, avšak na straně druhé prodlužuje jeho trvání [5]. Jako jedna z hlavních je vnímána vazba na plazmatický albumin [7]. Ten je zodpovědný za onkotický tlak plazmy, váže ligandy – volné mastné kyseliny, kalcium, některé hormony, bilirubin, transportuje měď, vitaminy, toxiny a léky [7]. Každá albuminová molekula má minimálně 6 vazebných míst pro farmaka a endogenní ligandy [7]. Dvě z nich mají velmi těsnou vazbu k volným mastným kyselinám, jedno pro bilirubin a dvě hlavní vazebná místa pro kyselá farmaka označovaná jako „site I“ a „site II“. Na „site I“ se váží farmaka jako warfarin a fenylbutazon, na „site II“ diazepam a ibuprofen. Především pro zásaditá léčiva je hlavním vazebným proteinem α1 kyselý glykoprotein (AGP) [7].
Množství transportních bílkovin je ovlivněno věkem, výživou a chronickými onemocněními, zejména záněty, infekcemi a nádory, kdy dochází ke zvýšení koncentrace AGP, a tím se snižuje volná frakce léčiv, jež se na tuto bílkovinu akutní fáze váží [7]. Změny jejich koncentrací (např. při hypoalbuminemii) mohou snížit koncentraci vázaného léčiva (inaktivní forma) a zvýšit koncentraci léčiva nevázaného. Množství aktivní, tzv. volné formy určí výsledný účinek daného léčiva. Změny koncentrací plazmatických bílkovin tedy mohou významně ovlivnit toxicitu léčiv, která jsou na transportní bílkoviny silně vázána nebo mají úzké terapeutické rozmezí (jako např. amiodaron, kys. valproová, fenytoin) [7]. Zvláště u starých nemocných je nutno myslet na stanovení volných frakcí těchto léčiv.
Uveďme příklad z praxe, kdy podávání nesteroidních antiflogistik (NSAID) může u pacienta výrazně zvýšit podíl volné frakce warfarinu, zvyšuje se tedy významně jeho účinnost. Lékaři však považují tuto kombinaci léčiv za kontraindikovanou proto, že obě léčiva, a to jak samotným mechanismem účinku warfarinu, tak nežádoucím účinkem NSAID, mohou zapříčinit vznik krvácení. Je však třeba myslet i na tento vzájemný interakční mechanismus, který zde hraje významnou roli.
Léková forma
Původní termín „biologická dostupnost“ označoval schopnost tablet, dražé a tobolek dostatečně rychle uvolňovat léčivou látku tak, aby byla k dispozici pro absorpci v trávicím ústrojí [2]. Dnes je tak označována farmaceutická dostupnost nebo liberace [2]. Liberace je zásadní především u tuhých lékových forem podávaných per os a per rectum. Rychlost, s jakou se léčivo uvolní z lékové formy, ovlivní koncentraci v místě vstřebávání, nástup účinku a délku jeho trvání [5].
Biologická dostupnost je pojem mnohem širší. Dříve se nízká biologická dostupnost řešila zvyšováním rozpustnosti úpravou účinné látky fyzikální či chemickou cestou, např. tvorbou solí, hydrátů, glykosylovaných derivátů, chelatací, lyofylizací, mikronizací či nanonizací [2]. V současné době se trendem stala příprava takových lékových forem, které zajišťují vyšší biologickou dostupnost a zároveň poskytují vhodnou stabilitu a aplikační komfort přípravku např. zvýšením smáčivosti, micelární solubilizací [2]. Jako příklad nám může posloužit použití enterosolventního obalu léčiva, který zajišťuje rozpad tablety až v neutrálním či mírně alkalickém prostředí tenkého střeva, nebo použití systému OROS (oral release osmotic system) u hydromorfonu. Při použití běžných lékových forem je k dosažení účinné analgezie nezbytné podávat hydromorfon každých 4−6 hodin, systémem OROS je zajištěna jeho účinná koncentrace po dobu 24 hodin [8].
Biologická dostupnost léčiva může být ovlivněna jedním faktorem nebo se může jednat o souhru více faktorů najednou, např. při podávání cyklosporinu A. U dříve používané lékové formy byla absorpce pomalá, neúplná a závislá na koncentraci žlučových kyselin ve střevě [9]. Novější lékové formy označované jako mikroemulze tyto parametry zlepšují. Současné požití tučného jídla nebo průjmové onemocnění snižují biologickou dostupnost, naopak požití před jídlem dostupnost zvyšuje [9].
Potrava
Potrava tedy může zasahovat nejen do rychlosti rozpadu lékové formy, uvolnění, rozpouštění, vstřebávání a průchodu léčiva trávicím traktem, ale její vliv je velice komplexní. Je mnoho léčiv, kde podání před jídlem (fenoxymetylpenicilin, levotyroxin, železo), s jídlem nebo po jídle (cefuroxim axetil, rivaroxaban) ovlivňuje jeho biologickou dostupnost. Některé složky potravy mohou vytvářet neabsorbovatelné nebo nerozpustné komplexy (např. tetracykliny a Ca2+) a tím snižovat biologickou dostupnost. V praxi klinického farmaceuta je na denním pořádku nutnost upozorňovat na obdobné konkrétní případy a upravovat zejména časový harmonogram podávání takových léků. Obzvlášť obtížná situace bývá u pacientů s polypragmazií.
Příkladem léčiva, jehož absorpci a potažmo účinek velmi ovlivňuje potrava, je levodopa, a to na podkladě kompetice o aktivní transportní mechanismus [2,10]. Na jedné straně stojí fakt, že bílkoviny a strava značně ovlivňují absorpci levodopy, a tím i její účinek (doporučeno užívat 15−20 minut před jídlem), na druhé straně je fakt, že pacient postižený Parkinsonovou chorobou je postižen i motorickou kolísavostí, a tedy je pro něj velmi důležitá dostatečná výživa, a to zejména bílkovinami [11]. Pokud by měl pacient nedostatek bílkovin, v budoucnu se jeho stav pravděpodobně zhorší, dieta s omezením bílkovin tedy není vhodným řešením. Vhodné je dobré načasování užívání léků a bílkovin. Bílkovinné jídlo je třeba podávat v době, kdy pomalá odezva na léčbu tolik nevadí (zejména večer) jako v aktivnější části dne [11]. V praxi to znamená, že snídaně a přesnídávka jsou složeny převážně ze sacharidů a tuků a koncentrované bílkovinné zdroje je vhodné zařadit do jídelníčku k večeru (maso, tvaroh, tvrdé sýry). Je třeba brát v potaz i samotné onemocnění, které je doprovázeno nepříjemnými symptomy (třes, špatná koordinace pohybů i při kousání, polykání, nauzea, ztráta chuti nebo čichu), při nekvalitní výživě může velmi snadno dojít k chřadnutí pacienta [11].
Uveďme další příklad z praxe: načasování současného podávání bisfosfonátu alendronátu (nalačno, 1 h před jídlem, zapít pouze vodou a být ve vzpřímené poloze), železa (rovněž nalačno, zapít kyselým nápojem) a levotyroxinu (rovněž nalačno). U tohoto konkrétního pacienta bylo doporučeno posunutí podávání železa na jinou denní dobu (poledne, večer) a současně pečlivá edukace o časovém harmonogramu užívání levotyroxinu a alendronátu v jednom dni v týdnu.
Komedikace
Na proces absorpce může mít vliv současné podávání více léčiv. Interakce mohou mít charakter buď synergický, nebo antagonistický, snížení biologické dostupnosti je mnohem častější než její zvýšení. Jedním typem interakcí jsou interakce na chemické úrovni, druhou skupinou jsou interakce vlastním farmakologickým účinkem (ovlivnění motility GIT, vazokonstrikce, vazodilatace apod.) [5].
Prostředí trávicího traktu pH žaludku
Hodnota pH žaludku se pohybuje obvykle v rozmezí 1,5−2,9 (nalačno), může stoupat až k hodnotám okolo 6,7 (po jídle). V duodenu se pH pohybuje v rozmezí 6,0−6,5, v jejunu okolo 6,8, v ileu až 7,4. V tlustém střevě nabývá hodnot 5,5−8, přičemž nižší pH se objevuje na začátku, je způsobeno vznikem kyselých fermentačních produktů bakteriální flóry [2]. Jako příklad ovlivnění účinnosti léčiv zde lze uvést nutnost vstřebání některých léčiv v dostatečně kyselém prostředí; změnou pH, tedy např. přidáním látek snižujících aciditu (velmi oblíbené inhibitory protonové pumpy), totiž může být narušena acidorezistentní léková forma a léčivo může být rozpuštěno již v žaludku, což může zabránit správnému vstřebání např. itrakonazolu. Správné načasování podání jednotlivých látek zde hraje velmi významnou roli. Pacienti užívající levotyroxin nebo přípravky s železem jsou leckdy správně informováni od předepisujícího lékaře o vlivu potravy na vstřebání léčiva, ovšem v nemocničním prostředí jsou jim do medikace přidána další léčiva, která mohou ovlivňovat jejich absorpci, a tedy je třeba zamýšlet se nad medikací pacienta komplexně, tj. dát do souvislosti jeho chronickou medikaci a medikaci podávanou v souvislosti např. s chirurgickým výkonem.
Motilita trávicího traktu
Lze říci, že vliv rychlosti vyprazdňování žaludečního obsahu na biologickou dostupnost je závislý na místě absorpce léčiva. U látek vstřebávajících se ze žaludku dochází ke zvýšení biologické dostupnosti se sníženou rychlostí vyprazdňování, zatímco léčiva absorbující se ve střevech vykazují naopak snížení biologické dostupnosti [2]. Vyprazdňování žaludku je spjato s příjmem potravy a je závislé především na objemu jídla (zvětšený objem zpočátku zvyšuje, později snižuje vyprazdňování žaludku), na složení potravy (tuky a bílkoviny zpomalují vyprazdňování žaludku), na viskozitě potravy (zvyšující se viskozita snižuje rychlost vyprazdňování), na teplotě stravy (teplá jídla urychlují vyprazdňování), na léčivých látkách (analgetika snižují, NaHCO3 nebo ranitidin zvyšují rychlost vyprazdňování žaludku) či alkoholu (konzumace snižuje rychlost vyprazdňování žaludku). Zpomalené vyprazdňování žaludku může být důsledkem některých onemocnění, často bývá narušena funkce nervů, které ovládají žaludeční svalovinu, jako např. při diabetes mellitus, Parkinsonově chorobě či sklerodermii (autoimunitní onemocnění pojivové tkáně s narušením nervosvalového přenosu v žaludku) [11].
Motilita střev pozitivně působí na rozpad kusových léčivých přípravků, rozpouštění a difuzi rozpuštěné léčivé látky směrem ke střevní membráně a ovlivňuje tak jeho absorpci [12]. S vyšší viskozitou nebo aplikací spasmolytik se motilita snižuje. Laxancia urychlují střevní pasáž a podstatně zkracují dobu kontaktu léčiv se sliznicí, mohou tedy absorpci snížit [12]. Zrychlení pasáže může mít vliv na absorpci retardovaných lékových forem, kdy množství takového vstřebaného léčiva závisí na době setrvání v GIT. Obdobně je tomu u prokinetik, která ovšem ovlivňují spíše proximální část GIT, nemívají tak výrazný vliv [12]. Je třeba upozornit na vliv podávání opioidů na snížení motility, a tím i na zadržování střevního obsahu, a tedy na možné zvýšení absorpce látek [5]. S tímto problémem se setkáváme často u pacientů léčených na onkologii, kde jsou podávány opioidy jak po delší dobu, tak ve vyšších dávkách. Motilita bývá ovlivněna věkem pacienta, u starších pacientů je snížena, je zvýšen kontaktní čas se sliznicí, což může být příčinou nežádoucích účinků některých léčiv. Většinou nejde o zásadní snížení či zvýšení absorbovaného podílu dané látky, ovšem může být ovlivněn nástup účinku (zpomalený nástup účinku u analgetik, antiflogistik) [5].
Metabolizace
Přítomnost enzymů
V odlišných částech GIT se vyskytuje rozdílné množství tekutin a trávicích enzymů, jejichž úkolem je natrávení základních složek potravy, jako jsou bílkoviny, sacharidy, tuky a nukleotidy. Léčiva na bázi proteinu a genetických materiálů (pro genovou terapii) nemohou být podávána perorálně, pokud není použita speciální technologie přípravy lékové formy, která jejich rozkladu zabraňuje [2].
First pass efekt
Část léčiva podaného perorálně není vůbec absorbována z důvodu chemické degradace, inaktivace díky vzniku vazeb nebo komplexů, mikrobiální biotransformace apod. Další část léčiva může být metabolizována v průběhu transportu přes stěnu GIT. Nezměněné léčivo, které se dostane do portální žíly, může být biotransformováno v játrech nebo navráceno zpět do střev biliární exkrecí [2]. Mezi léčiva s vysokým first pass efektem řadíme např. lipofilní betablokátory (např. metoprolol), blokátory kalciových kanálů, triazolam, midazolam, morfin, omeprazol, lovastatin, simvastatin [13]. Rovněž dabigatran je léčivo s velmi nízkou biologickou dostupností (F = 4 %). Jedná se o „dvojité“ proléčivo, které se enzymatickou přeměnou (hydrolytické štěpení katalyzované esterázou) po absorpci v trávicím traktu mění na aktivní metabolit dabigatran. Biologická přeměna dabigatran etexilátu na dabigatran začíná ve střevě, do portální žíly vstupuje proléčivo i aktivní látka. V játrech je přeměna dokončena (konjugace na farmakologicky aktivní acylglukuronid) a přibližně 20 % léčiva je vyloučeno biliárním systémem. K dosažení potřebných plazmatických koncentrací léčiva je tedy nutno podávat relativně vysoké dávky. Vstřebávání je velmi závislé na kyselém prostředí žaludku a tenkého střeva, je sníženo o 20−25 % u pacientů, kteří jsou léčeni inhibitory protonové pumpy [14].
Biorytmy
Téměř všechny fyziologické funkce, včetně absorpce léčiva, jsou závislé na čase, přičemž nejvýznamnější jsou výkyvy v průběhu dne (tzv. cirkadiánní rytmy) [2]. Prokázány byly u srdeční frekvence, tělesné teploty, krevního tlaku, prokrvení orgánů, plazmatické koncentrace hormonů, koncentrace neurotransmiterů a druhých poslů (např. kortizolu, melatoninu, inzulinu, prolaktinu apod.), množství kolujících červených a bílých krvinek a krevních destiček, funkce plic (minutová ventilace, usilovná vitální kapacita), jater (metabolismus, průtok krve, first pass efekt) a ledvin (glomerulární filtrace, pH a objem moči, vylučování elektrolytů), gastrointestinální motility, kyselosti žaludku, času potřebného k vyprázdnění žaludku a u řady dalších fyziologických funkcí. Rovněž nástup některých nemocí a jejich symptomů je závislý na denním rytmu, např. astmatické záchvaty jsou častější kolem 4. hodiny ranní nebo srdeční infarkty se nejčetněji vyskytují v ranních hodinách [2].
Intra a interindividuální variabilita organismů
U interindividuální variability organismů se z velké části jedná o geneticky podmíněné změny v metabolizaci léčivých látek (aktivita N acetyltransferázy, hydrolázy, genetický polymorfismus CYP2D6 apod.), na jejichž základě může být lidská populace rozdělena na tzv. rychlé, pomalé nebo intermediární metabolizátory [2]. Rovněž je zde třeba uvést vliv věku pacienta. U dětí může být biotransformace léčiv odlišná od dospělých. Ve stáří dochází ke zpomalování metabolismu některých léčiv snížením aktivity biotransformačních enzymů v játrech. Významně může být v důsledku sníženého průtoku přes játra snížen efekt prvního průchodu játry, a tedy zvýšena biologická dostupnost některých léčiv, u starších pacientů je proto doporučeno zahajovat léčbu těmito látkami třetinovou až poloviční dávkou oproti mladším pacientům. Dokumentována je rovněž snížená salivace, snížená střevní perfuze, klesá i počet receptorů pro aktivní transport látek [15].
K nejvýznamnějším interindividuálním rozdílům se řadí onemocnění (polymorbidita), uplatňují se poruchy funkce orgánů, jako jsou poruchy trávicího systému, kde místo absorpce může být poškozeno např. při zánětu střevní sliznice nebo přímo chyběním určité části traktu, změny ve vazbě léčiv nebo snížená funkce eliminačních orgánů, např. změna biologické dostupnosti některých léčiv u pacientů s renálním selháním [5]. Vedle těchto faktorů působí na biologickou dostupnost také vlivy vnějšího prostředí, mezi nimi stravovací návyky, kouření, lékové interakce a expozice toxickým látkám [2].
Závěr
Klinický význam biologické dostupnosti spočívá v tom, že umožní posoudit vztah mezi množstvím léčiva, které se nemocnému podává, a množstvím, které se skutečně může uplatnit ve farmakoterapeutickém účinku [1]. Protože koncentrace léčiva v krvi bývá v dynamické rovnováze s jeho efektivní koncentrací v místě účinku, pak údaje o biologické dostupnosti léčiva v dané lékové formě umožňují odvodit nástup, intenzitu a dobu působení léčiva. Vysoká presystémová eliminace (nízká absolutní biologická dostupnost) vyžaduje velký rozdíl v definovaném obsahu léčiva v lékové formě; to platí zejména pro perorální lékové formy např. u betablokátorů, nitrátů, salicylátů, analgetik narkotického typu [1]. Kvantitativní hledisko biologické dostupnosti musí být konstantní, aby lékař při neobvyklých a neočekávaných reakcích nemocného na léčivý přípravek mohl vyloučit nedostatečnou míru biologické dostupnosti jako zdroj komplikací. To platí i pro rychlostní hledisko [1].
Seznam použité literatury
- [1] wikiSkripta, Matematický popis farmakokinetických procesů, http://www.wikiskripta.eu/index.php
- [2] Vraníková B, Gajdziok J. Biologická dostupnost léčiva a možnosti jejího ovlivňování. Čes Slov Farm 2015; 64: 7–13.
- [3] Favea. Terapeutické systémy – řízené uvolňování léčiv z lékových forem: http://www.favea.cz/aktuality/terapeuticke systemy rizene uvolnovani leciv z lekovych forem drug delivery systems (navštíveno 10. 2. 2017).
- [4] wikiskripta, Celková anestetika, http://www.wikiskripta.eu/index.php/Celkov%C3%A1_anestetika_(farmakologie).
- [5] Dostálek M, a kol. Farmakokinetika Praha: Grada Publishing, 2006; str. 1–35, 83–91.
- [6] Ciferská H, Urbanová M. Biosimilars v terapii zánětlivých revmatických onemocnění. Klin Farmakol Farm 2015; 29: 124−128.
- [7] Adámek T, Paluch Z, Alušík Š. Bílkoviny krevní plazmy ve stáří a volné frakce léčiv. Čes Geriatr Rev 2008; 6: 257−262.
- [8] Urbánek K. Hydromorfon v lékové formě s prodlouženým uvolňováním aktivním osmotickým systémem OROS. Remedia 2007; 17: 502−503.
- [9] Fialová J. Imunosupresiva – 2. část. Čes Dermatovenerol 2012; 2: 38−41.
- [10] Grundmann M. Lékové interakce I. Int Med Prax 2000; 1: 40−41.
- [11] Chocenská E. Parkinsonova choroba a vliv bílkovin v potravě. Sestra 2010; 6: 2.
- [12] Vlček J, Fialová D a kol. Klinická farmacie I. Praha: Grada Publishing, 2010: 66−69.
- [13] Fialová D, Topinková E. Principy farmakoterapie ve vyšším věku – význam poznatků geriatrické farmakologie. Postgraduální medicína 2004; 6, Příl. 3.
- [14] Malý R. Dabigatran. Remedia 2008; 18: 331−332.
- [15] Wikipedia, Efekt prvního průchodu: https://cs.wikipedia.org/wiki/Efekt_prvn%C3%ADho_pr%C5