Farmakogenetika metabolismu léčiv – z výzkumu do klinické praxe
Souhrn:
Interindividuální variabilita lékové odpovědi je částečně způsobena genetickými rozdíly v metabolismu, částečně variabilitou citlivosti organismu na léky a částečně negenetickými faktory. Tento přehled shrnuje některé dědičné zdroje variability metabolismu léčiv, které mohou ovlivnit individuální reakci na léky, a to jak z hlediska léčebného, tak nežádoucího účinku. V přehledu uvádíme některé důležité aspekty této oblasti s důrazem na polymorfní enzymy I. a II. fáze metabolismu léčiv. Popsána jsou také doporučení pro testování některých genů zapojených do metabolismu léčiv.
Key words: polymorphism, drug metabolism enzymes, pharmacogenetics.
Summary:
Patients vary in their response to medicines. The variation between individuals partly arises due to the variability in drug metabolism enzymes, partly due to the variability in pharmacodynamics, and partly due to non genetic factors. This review summarizes inherited differences in drug metabolism which can affect individual responses to drugs, both in terms of therapeutic effect as well as adverse effects. The importance of the polymorphisms in phase I and phase II of drug metabolism is highlighted. Recent recommendations for testing of some genes involved in drug metabolism are also described.
Úvod
Výběr vhodného léčiva a jeho dávkování jsou často empirické, opírají se o anamnézu, klinické a laborato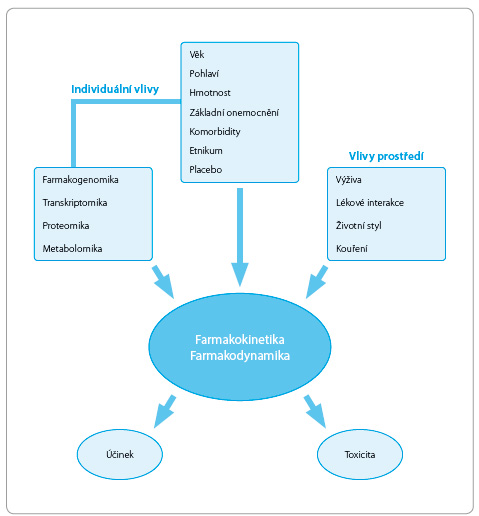 rní vyšetření a o poznatky o působení léčiva. Při dosavadním postupu se setkáváme s výraznou variabilitou klinického účinku. Někdy nemá léčba žádoucí léčebný efekt a u některých nemocných může být provázena nežádoucími účinky. Uspokojivá odpověď na léčbu většiny běžných onemocnění nastupuje u 30–60 % nemocných. V některých klinických situacích jsou odchylky ještě výraznější. Například nesteroidní antiflogistika ze skupiny koxibů účinkují až u 80 % léčených, ale průměrná účinnost protinádorové chemoterapie je pouze 25 %. Také bezpečnost léčby se liší v závislosti na daném onemocnění a použitém léčivu. Někdy jsou nežádoucí účinky tak závažné, že představují bezprostřední příčinu hospitalizace nebo vedou ke zrušení registrace léčiva [1].
rní vyšetření a o poznatky o působení léčiva. Při dosavadním postupu se setkáváme s výraznou variabilitou klinického účinku. Někdy nemá léčba žádoucí léčebný efekt a u některých nemocných může být provázena nežádoucími účinky. Uspokojivá odpověď na léčbu většiny běžných onemocnění nastupuje u 30–60 % nemocných. V některých klinických situacích jsou odchylky ještě výraznější. Například nesteroidní antiflogistika ze skupiny koxibů účinkují až u 80 % léčených, ale průměrná účinnost protinádorové chemoterapie je pouze 25 %. Také bezpečnost léčby se liší v závislosti na daném onemocnění a použitém léčivu. Někdy jsou nežádoucí účinky tak závažné, že představují bezprostřední příčinu hospitalizace nebo vedou ke zrušení registrace léčiva [1].
Variabilita lékové odpovědi je vyvolána zejména základním onemocněním, popřípadě souběžně probíhajícím dalším onemocněním. Významně se uplatňuje porucha funkce trávicího systému, změny ve vazbě léčiv, poruchy hemodynamiky a snížená funkce eliminačních orgánů. Vedle těchto faktorů působí i vlivy vnějšího prostředí, např. stravovací návyky, kouření, lékové interakce a expozice toxickým látkám (obr. 1).
Biotransformace xenobiotik včetně léčiv se běžně dělí do dvou fází. V první fázi probíhají oxidace, převážně závislé na enzymatickém systému cytochromu P450, nebo další děje, jako jsou například redukce a hydrolýzy. V této fázi dochází zpravidla k zavedení polární skupiny do základní molekuly. Oxidace, hydroxylace, demethylace a další modifikace cizorodé látky vytvářejí produkty, které jsou polárnější a snadněji se vylučují ledvinami. Ve druhé fázi dochází ke konjugačním reakcím, např. s kyselinou glukuronovou, sírovou nebo s glycinem.
Při oxidační přeměně xenobiotik se v první fázi biotransformace výrazně uplatňuje systém cytochromu P450 katalyzující oxidaci celého spektra endogenních i exogenních substrátů. Zvyšuje se tím jejich rozpustnost ve vodě, což usnadňuje vylučování xenobiotik z organismu. Všechny popsané izoenzymy cytochromu P450 tvoří tzv. superrodinu, která je podle podobnosti aminokyselinové struktury rozdělena do dalších rodin a podrodin označených arabskými číslicemi a velkými písmeny.
Cytochrom P450 zodpovídá za metabolickou přeměnu více než tří čtvrtin léčiv. Na biotransformačních procesech se u člověka podílejí zejména izoenzymy CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1 a CYP3A4/5. Pro enzymy metabolizující léčiva je charakteristická různě široká substrátová specifita. Více izoenzymů katalyzuje jeden typ reakce, to umožňuje biotransformaci látky alternativní metabolickou cestou při absenci, inhibici nebo geneticky modifikované aktivitě hlavního metabolizujícího enzymu.
Význam farmakogenetiky
Interindividuální a intraindividuální rozdíly v metabolismu jsou zčásti ovlivněny faktory genetickými. Jejich podrobnějším studiem se zabývá farmakogenetika/farmakogenomika. Pojem farmakogenetika navrhl Friedrich Vogel v roce 1959 jako označení pro novou vědní disciplínu aplikující genetiku ve farmakologii. Koncept farmakogenetiky navázal na pozorování extrémních interindividuálních rozdílů v koncentraci léčiv v krvi po podání stejné dávky léčiva. Farmakogenetika sleduje jen omezené množství biomarkerů na úrovni DNA, zejména polymorfismy v genech, které působí na chemickou přeměnu léčiv v organismu. Farmakogenomika zkoumá souvislosti mezi variabilitou lékové odpovědi v kontextu celého genomu. Do farmakogenetiky/farmakogenomiky se obvykle nezařazuje zjišťování genetické výbavy pacienta, která ovlivňuje charakter a závažnost onemocnění, ačkoli i tato genetická výbava podstatně působí na úspěch nebo neúspěch farmakoterapie.
Z historického pohledu byla farmakogenetika studována nejprve v souvislosti s velkou fyziologickou variabilitou glukóza‑6‑fosfát dehydrogenázy, jejíž deficit se u nás většinou manifestuje jako hemolytická anémie zejména v souvislosti s podáváním některých antibiotik, kyseliny acetylsalicylové apod. Dalším krokem bylo v polovině minulého století rozpoznání významu enzymu N‑acetyltransferázy 2, který je příčinou odlišné reakce na isoniazid a sulfonamidy. Následoval objev polymorfismu CYP2D6 v souvislosti s biotransformací debrisochinu a sparteinu.
Genetická rozmanitost je způsobena výskytem genetických variant, které jsou představovány především záměnami jednotlivých nukleotidů. V lidském genomu se předpokládá existence několika milionů genetických variant, z nichž většina nemá funkční vliv na kódovaný protein. Nicméně v regulačních i kódujících částech genů se vyskytuje množství variant ovlivňujících koncentrace daného proteinu nebo měnících aminokyselinové složení, které je významné pro aktivitu enzymu.
Variabilita metabolické aktivity je dána různým typem alel (jedna z více alternativních forem genu) daného enzymu. Někteří jedinci mají alely, jež zodpovídají za rychlý nebo ultrarychlý metabolismus substrátů daného enzymu (rychlí metabolizátoři, extensive metabolizers, fenotyp EM, nebo ultrarychlí metabolizátoři, ultra‑extensive metabolizers, fenotyp UM), u jiných se naopak vyskytují alely, které způsobují snížení metabolické kapacity enzymu (pomalí metabolizátoři, poor metabolizers, fenotyp PM). Heterozygoti mají metabolickou aktivitu buď normální, nebo jen lehce sníženou (normální, EM, nebo intermediární metabolizátoři, fenotyp IM).
Pokud se v populaci vyskytuje některá z genomových variant metabolizujícího enzymu u více než 1 % jedinců, označujeme enzym jako polymorfní. Genetické polymorfismy se týkají genů pro enzymy metabolizující léčiva, ale i genů kódujících lékové přenašeče či receptory. V důsledku polymorfismů mohou být ovlivněny farmakokinetické i farmakodynamické děje v organismu. Biotransformační změny mohou vést k bioaktivaci nebo k biodegradaci léčiva. Při bioaktivaci se původně neúčinná látka, označovaná jako proléčivo (prodrug), přeměňuje na účinnou látku, při biodegradaci se přeměňuje léčivo na farmakologicky neúčinné metabolity. V tomto pojetí je třeba chápat i význam polymorfismů typu EM a PM.
V tomto přehledu se soustřeďujeme na geneticky podmíněné změny metabolismu léčiv, které se mohou projevit změnou odpovědi na léčbu nebo výskytem nežádoucích účinků.
Izoenzym CYP2D6
Relativní množství izoenzymu CYP2D6 představuje asi 2 % ze všech enzymů cytochromu P450. Nicméně je jeho významnou součástí, protože katalyzuje oxidaci 20–30 % běžně užívaných léčiv. Spektrum jeho substrátů je velmi široké a zahrnuje psychofarmaka, lipofilní beta‑blokátory, některá antiarytmika a opioidy. Gen CYP2D6 je vysoce polymorfní, bylo popsáno více než 120 variantních alel, jejichž frekvence výskytu se liší podle rasové příslušnosti. U pomalých metabolizátorů převažuje tendence ke kumulaci léčiva, u ultrarychlých metabolizátorů naopak dochází ke snižování plazmatických koncentrací aktivního léčiva pod terapeuticky účinné rozmezí.
Mezi nežádoucí účinky substrátů CYP2D6 u jedinců s variantní alelou nesoucí fenotyp PM se řadí např. nedostatečná biotransformace a následná kardiotoxicita tricyklických antidepresiv, arytmie po podávání antiarytmik a výrazná bradykardie při léčbě beta‑blokátory. Snížené až chybějící analgetické působení codeinu u jedinců s fenotypem PM je důsledkem nedostatečné tvorby aktivního metabolitu. Zvláštní riziko při podávání codeinu u pacientů s fenotypem UM představuje potenciální kumulace morfinu (aktivního metabolitu codeinu), což bylo pozorováno např. u kojenců matek, které užívaly vyšší dávky codeinu.
Význam částečného či úplného deficitu aktivity CYP2D6 pro klinickou praxi potvrzují výsledky vlastního pozorování [2]. Stanovovali jsme genotyp CYP2D6 u 60 nemocných, kteří byli na našem pracovišti geneticky vyšetřeni pro projevy nežádoucích účinků léčiv. Kontrolní skupinu jsme vytvořili z 218 zdravých nepříbuzných dobrovolníků. V souboru pacientů bylo v porovnání s kontrolní skupinou signifikantně méně rychlých metabolizátorů CYP2D6 (25,0 % vs. 49,8 %), a naopak zastoupení pacientů s parciálním deficitem enzymatické cesty bylo vyšší v porovnání s kontrolní skupinou (58,3 % vs. 38,5 %). Rovněž pomalých metabolizátorů CYP2D6 bylo ve skupině nemocných více než ve zdravé populaci (13,3 % vs. 6,8 %).
V součas nosti se upozornění na klinický význam variability CYP2D6 stalo u některých léčiv součástí souhrnu údajů o léčivém přípravku (SPC), tab. 1. Při volbě těchto léčiv je třeba v případě znalosti fenotypu UM či PM věnovat pozornost úpravě dávkování a pečlivému sledování klinické odpovědi na léčbu. Při atypické odpovědi na léčbu těmito látkami lze potom zvažovat variabilitu CYP2D6 jako jednu z možných příčin. Aktivita enzymu CYP2D6 může ovlivnit též osud aktivních metabolitů některých léčiv. Příkladem je tamoxifen, metabolizovaný převážně CYP3A4 na N‑demethyl tamoxifen, který se následně přeměňuje pomocí CYP2D6 na jiný aktivní metabolit endoxifen. U pacientů s nízkou aktivitou enzymu CYP2D6 jsou koncentrace endoxifenu přibližně o 75 % nižší než u pacientů s normální aktivitou CYP2D6. Podávání silných inhibitorů CYP2D6 snižuje cirkulující hladiny endoxifenu v podobném rozsahu.
nosti se upozornění na klinický význam variability CYP2D6 stalo u některých léčiv součástí souhrnu údajů o léčivém přípravku (SPC), tab. 1. Při volbě těchto léčiv je třeba v případě znalosti fenotypu UM či PM věnovat pozornost úpravě dávkování a pečlivému sledování klinické odpovědi na léčbu. Při atypické odpovědi na léčbu těmito látkami lze potom zvažovat variabilitu CYP2D6 jako jednu z možných příčin. Aktivita enzymu CYP2D6 může ovlivnit též osud aktivních metabolitů některých léčiv. Příkladem je tamoxifen, metabolizovaný převážně CYP3A4 na N‑demethyl tamoxifen, který se následně přeměňuje pomocí CYP2D6 na jiný aktivní metabolit endoxifen. U pacientů s nízkou aktivitou enzymu CYP2D6 jsou koncentrace endoxifenu přibližně o 75 % nižší než u pacientů s normální aktivitou CYP2D6. Podávání silných inhibitorů CYP2D6 snižuje cirkulující hladiny endoxifenu v podobném rozsahu.
Izoenzym CYP2C9
Enzym CYP2C9 je nejvíce zastoupeným peptidem cytochromu P450, představuje 30 % jeho celkového množství. Je významný pro biotransformaci asi 10 % běžně užívaných léčiv. Podílí se např. na metabolismu nesteroidních antiflogistik, antiepileptik, antikoagulancií a antihypertenziv. Na snížení enzymové aktivity se nejčastěji podílejí alely CYP2C9*2 a CYP2C9*3. Výskyt variantních alel se výrazně liší podle rasové příslušnosti jedince. V bělošské (europoidní) populaci se variantní alely vyskytují asi u jedné třetiny jedinců. Homozygoti CYP2C9*3 s nejnižší metabolickou aktivitou představují nejvýše 0,4 % jedinců.
Již řadu let se věnuje velká pozornost genetickým determinantám metabolismu a variability účinku a dávkování warfarinu. Odbourávání S‑warfarinu se uskutečňuje cestou CYP2C9, zatímco katabolismus R‑warfarinu prostřednictvím izoenzymů CYP1A2 a CYP2C19. Menší význam má CYP3A4, který se podílí na metabolismu obou izomerů. Vliv variant CYP2C9*2,*3 byl zkoumán v řadě studií. Přítomnost variantních alel vede k laboratorním ukazatelům předávkování a k prodloužení času do stabilizace INR (International Normalized Ratio). Zásadní úlohu v individuální, geneticky podmíněné citlivosti vůči warfarinu hraje též gen pro cílový enzym blokovaný warfarinem (VKORC1), který kóduje podjednotku 1 enzymového komplexu vitamin K‑epoxidreduktázy [4]. Americký úřad Food and Drug Administration (FDA) stratifikuje populaci na základě těchto dvou genů na normální, senzitivní a vysoce senzitivní respondéry s příslušnými doporučenými úpravami dávkování. Přestože některé menší studie přinesly rozporuplné výsledky týkající se přínosu úpravy dávkování warfarinu na základě farmakogenetických dat, nedávno publikovaná studie ENGAGE AF TIMI‑48, provedená v souboru 14 348 pacientů, prokázala, že variabilita v uvedených dvou genech významně určuje riziko krvácení během prvních 90 dní podávání warfarinu [5].
Kromě warfarinu je gen CYP2C9 významný také pro metabolismus losartanu a phenytoinu. Vzhledem k tomu, že tvorba aktivního metabolitu losartanu je pouze minoritní cestou biotransformace tohoto léčiva, změna jeho koncentrací se v přítomnosti variantních alel klinicky neprojevuje.
Větší klinický význam se přisuzuje genetickým faktorům ovlivňujícím farmakokinetiku phenytoinu. Pouze 5 % phenytoinu se vylučuje v nezměněné formě močí nebo stolicí. Některé práce ukazují na vliv farmakogenetických faktorů na koncentrace a dávkování phenytoinu. Uplatňují se variantní alely CYP2C9*2 a CYP2C9*3, které zodpovídají za 31 % variability plazmatických koncentrací phenytoinu. Ve skupině s variantními alelami jsou také o 30 % vyšší průměrné koncentrace léčiva ve srovnání s jedinci s fenotypem IM. Lze proto předpokládat, že u těchto nemocných se budou častěji vyskytovat nežádoucí účinky, jako je cefalea a nystagmus, které závisejí na vyšších koncentracích léčiva. Jsou známy též rozdíly ve výskytu kožních reakcí u jednotlivých alel. Jednoznačnou klinickou interpretaci farmakogenetických vlivů znesnadňuje složitý metabolismus phenytoinu. Stanovování plazmatických koncentrací phenytoinu je v současnosti běžnou součástí terapeutického monitorování, určování přítomnosti variantních alel se proto běžně neprovádí.
Izoenzym CYP2C19
Izoenzym CYP2C19 představuje asi 3% podíl všech enzymů cytochromu P450. Jeho pol ymorfismus byl popsán v souvislosti se schopností jedinců metabolizovat S‑mephenytoin, což umožnilo jejich fenotypové rozlišení na PM a EM. Podíl jedinců s fenotypem PM v populaci se liší podle etnika; fenotyp PM se vyskytuje asi u 20 % příslušníků asijské populace a vzácnější (2–3 %) je u bělošské populace. V současnosti je popsáno více než 35 bodových mutací a přibližně u jedné třetiny z nich je znám vliv na aktivitu enzymu. Většinou se jedná o snížení aktivity, pouze variantní alela s tradičním označením CYP2C19*17 aktivitu enzymu zvyšuje. Mezi nejčetnější variantní alely se řadí CYP2C19*2 a *3, kde dochází k tvorbě zkrácených proteinů a ke snížení metabolické aktivity. Mezi substráty patří několik farmakoterapeutických skupin (tab. 2): inhibitory protonové pumpy, antidepresiva, anxiolytika, antimykotika a léčiva bránící agregaci trombocytů.
ymorfismus byl popsán v souvislosti se schopností jedinců metabolizovat S‑mephenytoin, což umožnilo jejich fenotypové rozlišení na PM a EM. Podíl jedinců s fenotypem PM v populaci se liší podle etnika; fenotyp PM se vyskytuje asi u 20 % příslušníků asijské populace a vzácnější (2–3 %) je u bělošské populace. V současnosti je popsáno více než 35 bodových mutací a přibližně u jedné třetiny z nich je znám vliv na aktivitu enzymu. Většinou se jedná o snížení aktivity, pouze variantní alela s tradičním označením CYP2C19*17 aktivitu enzymu zvyšuje. Mezi nejčetnější variantní alely se řadí CYP2C19*2 a *3, kde dochází k tvorbě zkrácených proteinů a ke snížení metabolické aktivity. Mezi substráty patří několik farmakoterapeutických skupin (tab. 2): inhibitory protonové pumpy, antidepresiva, anxiolytika, antimykotika a léčiva bránící agregaci trombocytů.
Zvýšená pozornost byla věnována problematice inhibitorů protonové pumpy. Jsou to proléčiva vyžadující přeměnu v kyselém prostředí intracelulárních kanálků parietálních buněk na aktivní látku, která má schopnost blokovat sekreci žaludeční kyseliny parietální buňkou. Neaktivní metabolity inhibitorů protonové pumpy vznikají po biotransformaci, na které se podílí zejména enzym CYP2C19 a v malé míře také CYP3A4. Deficit CYP2C19 pak výrazně zpomaluje odbourávání omeprazolu a zvyšuje jeho léčebný účinek. U nemocných s funkčním deficitem CYP2C19 je při běžném dávkování výrazněji potlačena žaludeční acidita.
Až sedminásobně zvýšené plochy pod křivkou plazmatické koncentrace (area under curve, AUC) omeprazolu jsou nacházeny u pomalých metabolizátorů po podání prvních dávek ve srovnání s rychlými metabolizátory. Tento rozdíl se však snižuje při opakovaném podávání, protože u rychlých metabolizátorů dochází k autoinhibici omeprazolem a k částečné kumulaci léku. Autoinhibice se však nemůže projevit u pomalých metabolizátorů kvůli nepřítomnosti funkčního enzymu. Mezi inhibitory protonové pumpy existují značné rozdíly v biotransformaci i při účasti stejného izoenzymu cytochromu P450 [6]. Autoinhibice se neuplatňuje u lansoprazolu ani u pantoprazolu. Zvýšení AUC rabeprazolu je u pomalých metabolizátorů méně výrazné.
Důležité jsou terapeutické aspekty působení uvedených genetických odchylek. Inhibitory protonové pumpy mají obecně dobrou toleranci, proto se geneticky podmíněné zvýšené koncentrace neprojevují zvýšenou toxicitou a úprava dávkování není nutná. Při hodnocení eradikačních režimů byl sledován také léčebný účinek. Výsledky klinického hodnocení dokumentují nižší rychlost a menší úspěšnost eradikace u pacientů s fenotypem EM ve srovnání s fenotypem PM. Genetický polymorfismus CYP2C19, ovlivňující metabolismus inhibitorů protonové pumpy, může obdobnými mechanismy ovlivnit také riziko lékových interakcí, např. snížení účinnosti protidestičkového léčiva clopidogrelu u pacientů užívajících inhibitory protonové pumpy; toto riziko je vyšší u pomalých metabolizátorů v důsledku vyšších koncentrací omeprazolu.
Izoenzym C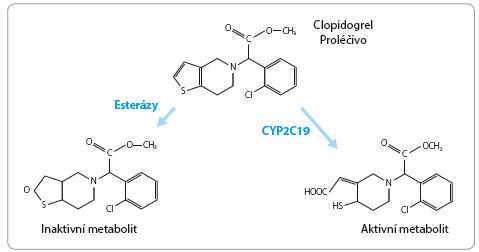 YP2C19 se také významně podílí na konverzi proléčiva clopidogrelu na aktivní metabolit, který inhibuje agregaci destiček prostřednictvím ireverzibilní vazby na receptory P2Y12. Alela ztráty funkce, označovaná jako CYP2C19*2, vede ke snížené produkci aktivního metabolitu clopidogrelu a je spojena se zvýšeným rizikem kardiovaskulárních příhod u nemocných po perkutánní koronární intervenci. Alela CYP2C19*17 predikuje vyšší aktivitu enzymu i tvorbu aktivního metabolitu ve srovnání s nosiči alely CYP2C19*1 (EM). Řada studií naznačuje, že antitrombotický účinek clopidogrelu je u pomalých metabolizátorů snížen až o 50 % ve srovnání s rychlými metabolizátory (obr. 2). Farmakogenetické monitorování vedoucí ke zvýšení účinnosti léčby clopidogrelem se stalo součástí nových doporučení [7,8].
YP2C19 se také významně podílí na konverzi proléčiva clopidogrelu na aktivní metabolit, který inhibuje agregaci destiček prostřednictvím ireverzibilní vazby na receptory P2Y12. Alela ztráty funkce, označovaná jako CYP2C19*2, vede ke snížené produkci aktivního metabolitu clopidogrelu a je spojena se zvýšeným rizikem kardiovaskulárních příhod u nemocných po perkutánní koronární intervenci. Alela CYP2C19*17 predikuje vyšší aktivitu enzymu i tvorbu aktivního metabolitu ve srovnání s nosiči alely CYP2C19*1 (EM). Řada studií naznačuje, že antitrombotický účinek clopidogrelu je u pomalých metabolizátorů snížen až o 50 % ve srovnání s rychlými metabolizátory (obr. 2). Farmakogenetické monitorování vedoucí ke zvýšení účinnosti léčby clopidogrelem se stalo součástí nových doporučení [7,8].
V současnosti se upozornění na význam polymorfní aktivity CYP2C19 týká řady léčiv a je již součástí SPC (tab. 2).
Izoenzymy CYP3A4/5
Nejdůležitější úlohu z celého systému cytochromu P450 představují izoenzymy CYP3A4/5, které metabolizují řadu xenobiotik i endogenních substrátů. Tyto izoenzymy představují asi 30 % celkového cytochromu P450 a zabezpečují přibližně polovinu celkové metabolické aktivity. Tento systém se podílí na metabolismu asi 150 běžně užívaných léčiv. Genový polymorfismus CYP3A4/5 se výrazněji neuplatňuje. K variabilitě jeho funkce podstatně přispívá indukce a inhibice vyvolaná léčivy a složkami potravy.
Genetický polymorfismus II. fáze biotransformačních procesů má význam pro vyloučení polárních látek z organismu.
N‑acetyltransferáza
Polymorfismus N‑acetyltransferázy 2 (NAT2) je znám od poloviny minulého století, zatímco průkaz polymorfismu N‑acetyltransferázy 1 (NAT1) je podstatně mladší. Genetický polymorfismus NAT2 je příčinou odlišné reakce na isoniazid. Metabolická aktivita rozlišila pomalé a rychlé acetylátory. Pomalí acetylátoři představují asi 60 % bělošské populace. Molekulární podstatou polymorfismu je přítomnost více než 15 alelických forem, z nichž většina enzymovou aktivitu snižuje. Tito pacienti mají vyšší koncentrace mateřské látky a jsou vystaveni zvýšenému riziku hepatotoxicity vyvolané isoniazidem a zvýšenému riziku periferní neuropatie. Pomalí acetylátoři jsou také ohroženi zvýšeným nebezpečím karcinogenního působení aromatických aminů.
Thiopurin‑S‑methyltransferáza
Detekce aktivity thiopurin‑S‑methyltransferázy (TPMT), cytozolického enzymu II. fáze biotransformace, je významná pro záchyt deficitní metabolické přeměny imunosupresiv azathioprinu a 6‑thioguaninu. Aktivitu TPMT je možné predikovat z výsledků genotypizace, nebo přímo sledovat rychlost konverze 6‑mercaptopurinu in vitro. Oba postupy, vycházející z jednoho odběru krve, umožňují identifikovat rizikové pacienty před zahájením léčby. Vyšetření aktivity TPMT je v současnosti nejčastějším screeningovým farmakogenetickým vyšetřením metabolismu léčiv v klinické praxi prováděným u nás i v zahraničí. Přibližně u 0,3 % naší populace je enzymová aktivita TPMT nedetekovatelná a u 11 % jedinců je aktivita výrazně snížená.
Nemocní s deficitem TPMT jsou ohroženi vysokým rizikem myelosuprese i rozvojem druhotné malignity vznikající v důsledku cytotoxického působení těchto léčiv. Příčinou je metabolická konverze proléčiva azathioprinu, při níž se většina podané dávky inaktivuje prostřednictvím TPMT na neaktivní metabolit, zatímco na aktivní cytotoxické a imunosupresivní m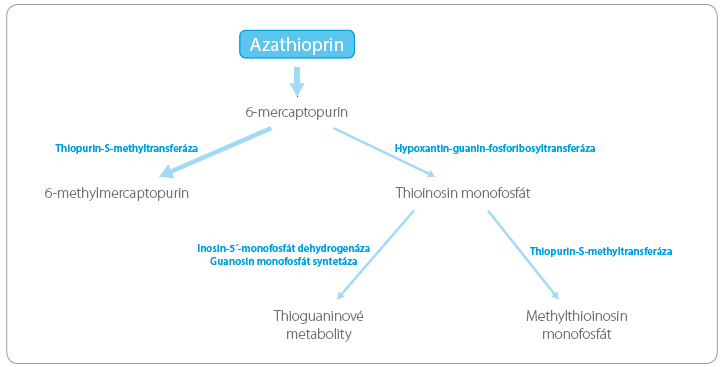 etabolity (thioguaninové metabolity) se přemění jen malá část dávky (obr. 3). Při deficitu TPMT dochází ke kumulaci cytotoxických metabolitů v cílových tkáních. I v našich podmínkách byly popsány významné polékové reakce v podobě pancytopenií spojených s deficitem TPMT, které minimálně v jednom případě skončily úmrtím nemocného [9].
etabolity (thioguaninové metabolity) se přemění jen malá část dávky (obr. 3). Při deficitu TPMT dochází ke kumulaci cytotoxických metabolitů v cílových tkáních. I v našich podmínkách byly popsány významné polékové reakce v podobě pancytopenií spojených s deficitem TPMT, které minimálně v jednom případě skončily úmrtím nemocného [9].
V souladu s literárními poznatky doporučujeme provádět toto vyšetření jako prevenci závažných projevů lékové toxicity. Určování genotypu/fenotypu TPMT jako screening před podáním azathioprinu vede k významnému snížení rizika myelotoxicity [10]. Podle závažnosti metabolického deficitu je možná úprava dávkování, případně volba jiného léčiva. Současné rutinní sledování klinické a laboratorní snášenlivosti léčby je ale nutné.
Uridindifosfát glukuronosyltransferáza
Genetický polymorfismus se významně projevuje také v případě genu uridindifosfát glukuronosyltransferázy 1A1 (UGT1A1). Nejčastěji se vyskytující molekulárněgenetický podklad je na úrovni promotoru genu označovaného jako UGT1A1*28. Homozygotní genotyp pro variantní alelu bývá často diagnostikován na základě fenotypových projevů jako familiární hyperbilirubinemie, vyskytující se v naší populaci přibližně s 12% frekvencí. Nosiči variantní alely jsou více ohroženi potenciální toxicitou navozenou užíváním významného cytostatika irinotecanu, inhibujícího topoizomerázu 1, které je používáno v léčbě kolorektálního karcinomu. Při metabolické přeměně irinotecanu vzniká účinný metabolit SN‑38, který je dále metabolizován na neaktivní a netoxické sloučeniny pomocí enzymu UGT1A1. Zpomalení glukuronidace tak souvisí se zvýšeným rizikem gastrointestinální toxicity a myelotoxicity.
Dihydropyrimidin dehydrogenáza
Význam deficitu dihydropyrimidin dehydrogenázy (DPD) se uvádí do souvislosti s toxicitou capecitabinu a 5‑fluorouracilu. Capecitabin je prekurzorem cytotoxicky působícího 5‑fluorouracilu, který se dále přeměňuje působením DPD na méně toxické produkty. Genetický deficit DPD proto může vést ke zvýšené toxicitě tohoto protinádorového léčiva. V genu DPD existuje mnoho desítek známých polymorfismů, což významně znesnadňuje predikci aktivity na základě znalosti genotypu. V praxi se proto upřednostňuje stanovení fenotypové aktivity tohoto enzymu, což dle publikovaných prací může při provedení před zahájením léčby vést ke snížení rizika toxicity při zachování účinnosti léčby, jsou‑li dávky léčiva upraveny dle výsledku vyšetření.
Využití farmakogenetiky v klinické praxi
Velké množství farmakogenetických poznatků popisujících variabilitu farmakokinetických procesů je často v rozporu s jejich uplatněním v klinické praxi. Tyto nedostatky se snaží překonat společné úsilí průmyslu, akademických institucí i klinických pracovišť. Také aktivity regulačních autorit svědčí o aktuálnosti této problematiky [11]. Zvyšuje se počet léčiv, jejichž souhrn údajů o přípravku obsahuje informaci o farmakogenomických biomarkerech. V roce 2014 uvádí databáze FDA tento údaj již přibližně u 100 registrovaných léčiv [3]. Většina doporučovaných farmakogenomických biomarkerů v metabolismu léčiv přispívá k omezení výskytu nežádoucích polékových reakcí. Širší uplatnění farmakogenetiky v oblasti biotransformace léčiv vyžaduje zavádění nových metod fenotypizace a genotypizace, které hodnotí aktivitu enzymů a přispívají ke zvýšení jejich dostupnosti v praxi. Při genotypizaci je metabolická aktivita odhadována z genetické výbavy jedince, při fenotypizaci určitého léčiva se přímo stanovují koncentrace mateřské látky a příslušného metabolitu [12]. Nutné jsou i dobře naplánované farmakogenetické studie provedené na dostatečně velkých souborech nemocných. Jedině komplexní přístup analýzy genetické variability metabolismu léčiv, prováděný společně se studiem geneticky podmíněných molekulárních znaků onemocnění, může přispět ke zvýšení bezpečnosti a účinnosti farmakoterapie.
Seznam použité literatury
- [1] Regierer B, Zazzu V, Sudbrak R, et al. Future of medicine: models in predictive diagnostics and personalized medicine. Adv Biochem Eng Biotechnol 2013; 133: 15–33.
- [2] Slanař O, Dražďáková M, Babiárová K, et al. Genotypizace cytochromu P450 2D6 a 2C19. Čas Lék čes 2007; 146: 708–711.
- [3] Pharmacogenomic Biomarkers in Drug Labeling FDA. http://www.fda.gov/Drugs/ScienceResearch/ResearchAreas/Pharmacogenetics/ucm083378.htm (navštíveno 12. 6. 2015).
- [4] Verhoef TI, Redekop WK, Daly AK, et al. Pharmacogenetic guided dosing of coumarin anticoagulants: algorithms for warfarin, acenocoumarol and phenprocoumon. Br J Clin Pharmacol 2014; 77: 626–641.
- [5] Mega LJ, Walker JR, Ruff CT, et al. Genetics and the clinical response to warfarin and edoxaban: findings from the randomised, double blind ENGAGE AF TIMI 48 trial. Lancet 2015; 385: DOI http://dx.doi.org/10.1016/S0140 6736(14)62219 4.
- [6] Hagymási K, Müllner K, Herszényi L, et al. Update on the pharmacogenomics of proton pump inhibitors. Pharmacogenomics 2011; 12: 873–888.
- [7] Price MJ, Berger PB, Teirstein PS, et al. Standard vs high dose clopidogrel based on platelet function testing after percutaneous coronary intervention: the GRAVITAS randomized trial. JAMA 2011; 305: 1097–1105.
- [8] Scott SA, Sangkuhl K, Stein CM, et al. Clinical Pharmacogenetics Implementation Consortium Guidelines for CYP2C19 Genotype and Clopidogrel Therapy: 2013 Update. Clin Pharmacol Ther 2013; 94: 317–323.
- [9] Slanař O, Chalupná P, Novotný A, et al. Fatal myelotoxicity after azathioprine treatment. Nucleosides, Nucleotides and Nucleic Acids 2008; 27: 661–665.
- [10] Colombel JF, Ferrari N, Debuysere H, et al. Genotypic analysis of thiopurine s methyltransferase in patients with Crohn‘s disease and severe myelosuppression during azathioprine therapy. Gastroenterology 2000; 118: 1025–1030.
- [11] Ehmann F, Caneva L, Papaluca M. European Medicines Agency initiatives and perspectives on pharmacogenomics. Br J Clin Pharmacol 2014; 77: 612–617.
- [12] Světlík S, Hronová K, Slanař O. Fenotypizace enzymů podílejících se na metabolismu léčiv. Čes slov Farm 2012; 61: 115–126.