Inhibitory SGLT2 (3. část) – glukuronidace jako hlavní cesta biotransformace gliflozinů
Souhrn:
Suchopár J. Inhibitory SGLT2 (3. část) – glukuronidace jako hlavní cesta biotransformace gliflozinů. Remedia 2022; 32: 312–316.
Pochopení cest metabolizace inhibitorů SGLT2 (gliflozinů) je jednou z podmínek bezpečné terapie těmito léčivy. Glifloziny se v organismu metabolizují prakticky jen glukuronidací, což je velmi neobvyklé a lékaři i farmaceuti s tím musejí počítat. Největší část všech gliflozinů se metabolizuje cestou UGT1A9, v případě dapagliflozinu je tato cesta rozhodující, naopak empagliflozin je metabolizován hned čtyřmi izoenzymy UGT, což se projevuje zanedbatelným vlivem jejich polymorfismů a lékových interakcí. Glukuronidy vzniklé metabolizací gliflozinů jsou dobře rozpustné ve vodě, a jsou proto eliminovány ledvinami aktivním transportem. Míra glukuronidace jednotlivých gliflozinů je různá, nejnižší je v případě empagliflozinu a nejvyšší u dapagliflozinu.
Summary:
Suchopar J. SGLT2 inhibitors (part 3) – glucuronidation as a major pathway for gliflozin biotransformation. Remedia 2022; 32: 312–316.
Understanding the metabolism pathways of SGLT2 inhibitors (gliflozins) is one of the conditions for safe therapy with these drugs. Gliflozins are metabolized in the body practically only by glucuronidation, which is very unusual. Both doctors and pharmacists must count it in. Most gliflozins are metabolized by UGT1A9. In the case of dapagliflozin, this is the decisive pathway, whereas empagliflozin is metabolized by four UGT isoenzymes, which manifests as a negligible effect of their polymorphisms and drug interactions. Glucuronides formed by the metabolism of gliflozins are readily soluble in water and are therefore eliminated by active renal transport. The degree of glucuronidation of individual gliflozins is different; the lowest is in the case of empagliflozin and the highest in the case of dapagliflozin.
Key words: gliflozins, SGLT, UDP‑glucuronosyltransferase, UGT1A9, genetic polymorphism, inhibition, induction.
Úvod
Hlavní cestou metabolizace gliflozinů je glukuronidace. Při ní vznikají dva hlavní metabolity gliflozinů, a to 3 O β glukuronidy (v případě sotagliflozinu), respektive směs 2 O β a 3 O β glukuronidů (v případě dapagliflozinu, kanagliflozinu, empagliflozinu a ertugliflozinu).
Ke glukuronidaci dochází
v poloze 2 nebo 3 „cukerné“ části molekuly gliflozinu
(obr. 1). Takto vzniklé metabolity jsou eliminovány močí
nebo žlučí.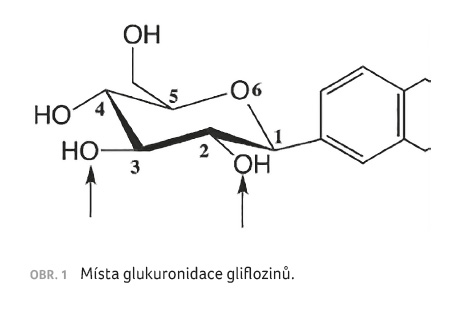
Shrnutí informací o glukuronidaci
gliflozinů
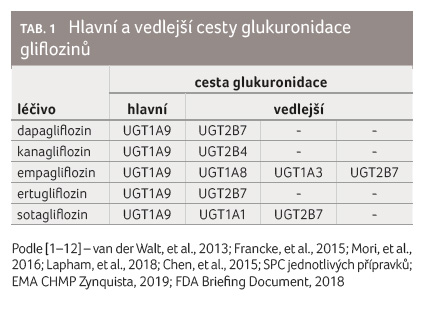
Glukuronidace gliflozinů probíhá převážně cestou uridindifosfát glukuronosyltransferázy UGT1A9, v menší míře se jí účastní také další izoenzymy, zejména UGT2B4, UGT2B7 a v případě empagliflozinu ještě minoritně UGT1A3 a UGT1A8 (tab. 1) [1−12]. Glukuronidace cestou UGT1A9 vede ke vzniku 3 O β glukuronidů. 2 O β glukuronidy vznikají především cestou UGT2B4 nebo UGT2B7. Glukuronidy gliflozinů nemají inhibiční účinek vůči SGLT2 (respektive SGLT1), a jsou proto farmakodynamicky neúčinné.
Glukuronidace dapagliflozinu
Dapagliflozin se glukuroniduje z přibližně 70 %, tj. nejvíce ze všech gliflozinů. Přitom hlavní cestou glukuronidace dapagliflozinu za vzniku 3 O β dapagliflozin glukuronidu je UGT1A9 a v případě minoritního 2 O β dapagliflozin glukuronidu je to UGT2B7. Glukuronidací dapagliflozinu vzniká zejména 3 O β dapagliflozin glukuronid a v mnohem menší míře (do 5 %) se tvoří 2 O β dapagliflozin glukuronid [13]. Také rozdíl v rychlosti tvorby 3 O β dapagliflozin glukuronidu in vitro je více než 40krát vyšší ve srovnání s 2 O β dapagliflozin glukuronidem (794 pmol min−1 mg−1 proteinu oproti 19,4 pmol min−1 mg−1 proteinu). Ke glukuronidaci dapagliflozinu dochází převážně v ledvinách. To vcelku není překvapivé, neboť aktivita UGT1A9 je největší právě v ledvinách, kde představuje kolem 50 % veškeré aktivity UGT [14]. Tvorba 3 O β dapagliflozin glukuronidu v mikrosomech ledvin byla třikrát vyšší než v mikrosomech jater a 109krát vyšší než v mikrosomech enterocytů [13]. To je velmi významné u pacientů s poruchou funkce ledvin, u nichž
se aktivita UGT1A9 snižuje.
Nadto bylo také prokázáno, že ve srovnání s pacienty s normální funkcí ledvin byly farmakodynamické účinky dapagliflozinu oslabeny poruchou funkce ledvin [13], neboť mírná, středně závažná a těžká porucha funkce ledvin vedla ke snížení clearance glukózy v ustáleném stavu o 42 %, 83 % a 84 %, tedy klinicky významně.
Nabízejí se otázky, zda blokáda glukuronidace dapagliflozinu bude mít vliv na míru expozice tomuto přípravku a na účinnost a bezpečnost takové případné souběžné terapie. Stejně tak se naskýtá otázka, zda genetický polymorfismus UGT1A9 může mít vliv na farmakokinetické vlastnosti, účinnost a bezpečnost terapie dapagliflozinem.
Odpověď na první otázku je k dispozici, neboť byla provedena studie zaměřená na lékovou interakci dapagliflozinu se selektivním inhibitorem UGT1A9 kyselinou mefenámovou [15]. V této studii u mladých zdravých dobrovolníků byla po dobu pěti dnů podávána kyselina mefenámová v dávkách 250 mg 4krát denně (úvodní dávka kyseliny mefenámové činila 500 mg). Před zahájením podávání kyseliny mefenámové a ve 2. dni jejího podávání ráno byl podán dapagliflozin v jednorázové dávce ve výši 10 mg. Vlivem kyseliny mefenámové došlo ke zvýšení plochy pod křivkou dapagliflozinu o 51 % (44–58 % na 95% hladině spolehlivosti [confidence interval, CI]) a jeho maximálních plazmatických koncentrací o 13 % (95% CI 3–24 %). Množství glukózy vyloučené močí za 24 hodin po podání dapagliflozinu bylo při podání s kyselinou mefenámovou oproti podání samotného dapagliflozinu vyšší v průměru o 18 %.
Také na druhou otázku je k dispozici odpověď. V říjnu 2021 byly publikovány výsledky studie [16] provedené se 187 diabetiky 2. typu se známými polymorfismy UGT1A9 (s2011404, rs6759892, rs75776477 a U923rs4 z U9G31rs4). Statistická analýza prokázala, že geometrický průměr poměru celkové clearance a dávky dapagliflozinu pro všechny studované polymorfismy UGT1A9 se nacházel v rozmezí hodnot divokého typu. Autoři proto výsledky shrnují tak, že studované polymorfismy UGT1A9 neměly žádný klinicky významný dopad na clearance dapagliflozinu ve vztahu k jeho dávce.
Glukuronidace kanagliflozinu
Kanagliflozin se glukuroniduje přibližně ze 34 %. Glukuronidací vznikají dva metabolity, a to 3 O β kanagliflozin glukuronid (označovaný také jako M7) a 2 O β kanagliflozin glukuronid (označovaný jako M5). Stejně jako v případě dapagliflozinu vzniká 3 O β kanagliflozin glukuronid cestou UGT1A9 a 2 O β kanagliflozin glukuronid cestou UGT2B4. Vliv genetického polymorfismu UGT1A9 a UGT2B4 byl zkoumán u 134 pacientů s diabetem 2. typu [2]. Bylo zjištěno, že polymorfismus UGT1A9*3 (rs72551330; p.Met33Thr) vyznačující se sníženou aktivitou UGT1A9 je spojen s 54% zvýšením expozice kanagliflozinu a polymorfismus UGT2B4*2, pro který je také charakteristické snížení aktivity, je spojen se zvýšením expozice kanagliflozinu o 18 %. Vliv genetického polymorfismu UGT1A9 byl potvrzen také v rozsáhlé studii zahrnující 1 616 probandů [17].
Kanagliflozin je in vitro inhibitorem UGT [18], přičemž k 50% inhibici glukuronidace propofolu, respektive 4 methylumbeliferonu (substráty UGT1A9) dochází již při koncentraci 2,9 μM, respektive 1,4 μM. Zda by kanagliflozin mohl být inhibitorem také in vivo, není dosud známo. Uvedené koncentrace kanagliflozinu v ledvinách jsou však patrně dosažitelné.
I když je podíl glukuronidace v případě kanagliflozinu menší než v případě dapagliflozinu, je přesto snížená aktivita UGT1A9, která je spojena s polymorfismem UGT1A9*3, důvodem klinicky relevantního zvýšení expozice kanagliflozinu. Nejsou známy dopady souběžného podávání selektivního inhibitoru UGT1A9 (kyseliny mefenámové) s kanagliflozinem, jsou však k dispozici výsledky studie, ve které byl spolu s kanagliflozinem podáván polyfunkční inhibitor UGT probenecid [19]. V této studii u zdravých dobrovolníků vedlo souběžné podávání obou léků ke zvýšení plochy pod křivkou kanagliflozinu o 21 % a k nárůstu jeho maximálních plazmatických koncentrací o 13 %. Tyto změny nejsou klinicky významné.
Na druhou stranu jsou k dispozici výsledky souběžného podávání kanagliflozinu s induktorem UGT1A9 a UGT2B4 rifampicinem [19]. V této studii u zdravých dobrovolníků vedlo souběžné podávání kanagliflozinu s rifampicinem ke snížení expozice kanagliflozinu o více než 50 %.
Glukuronidace empagliflozinu
Empagliflozin se glukuroniduje z přibližně až 28 %, tj. nejméně ze všech gliflozinů. Glukuronidací vznikají dva metabolity, a to 3 O β empagliflozin glukuronid (označovaný také jako M626/3), 2 O β empagliflozin glukuronid (označovaný jako M626/1) a 5 O empagliflozin glukuronid (označovaný také jako M626/2). Stejně jako v případě dapagliflozinu vzniká 3 O β empagliflozin glukuronid cestou UGT1A9, zatímco 2 O β empagliflozin glukuronid nebo 5 O empagliflozin glukuronid pak vznikají cestou UGT1A3, UGT1A8 a UGT2B7.
Podíl jednotlivých glukuronidů je následující [20]: nejvyšší podíl zaujímá 3 O β empagliflozin glukuronid, a to 12,3 %, následuje 2 O β empagliflozin glukuronid, jehož podíl činí 7,8 %, a poté 5 O empagliflozin glukuronid s podílem 2,1 %.
Nejsou k dispozici informace o vlivu genetického polymorfismu UGT1A9 ani o vlivu selektivního inhibitoru kyseliny mefenámové na farmakokinetické vlastnosti empagliflozinu. Dostupné jsou však informace o společné inhibici přenašeče organických aniontů OAT3 a UGT při souběžném podávání empagliflozinu a probenecidu (silný inhibitor OAT3 a polyfunkční inhibitor UGT), ve které vlivem probenecidu [21] došlo ke zvýšení plochy pod křivkou empagliflozinu o 53 % a ke zvýšení jeho maximálních plazmatických koncentrací o 26 %. Stejně tak jsou k dispozici výsledky studie, v níž byl podáván silný polyfunkční induktor UGT rifampicin. V této studii však byl rifampicin podán v jednorázové dávce (600 mg), což způsobilo na první pohled nepochopitelné zvýšení maximálních plazmatických koncentrací empagliflozinu o 75 % [21]. Mechanismem této lékové interakce však nebyla indukce UGT, ale jednalo se o inhibici OATP1B1 a OATP1B3, jejichž silným inhibitorem je rifampicin.
Glukuronidace ertugliflozinu
Ertugliflozin se glukuroniduje
z přibližně 43 %. Z výsledků studie in vitro
vyplývá [4], že hlavními metabolity ertugliflozinu jsou
3 O β ertugliflozin glukuronid (označovaný
jako M5c) a 2 O β ertugliflozin glukuronid
(označovaný jako M5a), první z uvedených metabolitů tvoří
kolem 80 %, druhý pak kolem 20 %. Přibližně 85 %
3 O β ertugliflozin-
-glukuronidu
vzniká cestou UGT1A9 a kolem 15 % pak cestou UGT2B7. Naopak
minoritní metabolit 2 O β ertugliflozin glukuronid
vzniká převážně (82 %) cestou UGT2B7 a jen minimálně
(19 %) cestou UGT1A9.
Pomocí farmakokinetického fyziologického modelování [22] byly zjišťovány možné změny farmakokinetických vlastností ertugliflozinu v případě souběžného podávání kyseliny mefenámové podávané v dávkách 500 mg 4krát denně. Vlivem kyseliny mefenámové by pravděpodobně došlo ke zvýšení plochy pod křivkou ertugliflozinu o 51 % (95% CI 48–54 %) a jeho maximálních plazmatických koncentrací o 19 % (95% CI 17–20 %).
Ve studii u zdravých dobrovolníků [23] byl prokázán indukční vliv podávání rifampicinu na glukuronidaci ertugliflozinu. Vlivem rifampicinu došlo ke snížení plochy pod křivkou ertugliflozinu o 39 % (90% CI 35–43 %), ke snížení jeho maximálních plazmatických koncentrací o 15 % (90% CI 3–26 %) a ke zkrácení průměrné hodnoty jeho biologického poločasu z 12,3 na 9,2 hodiny. Držitel rozhodnutí o registraci ertugliflozinu toto snížení expozice nepovažuje za klinicky relevantní, a proto nedoporučuje žádnou úpravu dávkování.
Glukuronidace sotagliflozinu
Sotagliflozin se glukuroniduje z přibližně 35 %. Hlavním metabolitem je 3 O β sotagliflozin glukuronid (označovaný též jako M19), který vzniká cestou UGT1A9. O metabolismu sotagliflozinu jsou dostupné jen kusé informace, a není proto známo, zda polymorfismus UGT1A9 ovlivňuje expozici tomuto přípravku či jeho účinnost a bezpečnost.
Držitelem rozhodnutí o registraci byly provedeny dvě studie, jejichž cílem bylo zjistit, zda inhibice, respektive indukce UGT1A9 vede ke klinicky významným změnám v expozici sotagliflozinu. V první studii u zdravých dobrovolníků (Study INT14937) byla v první fázi podána jednorázová dávka sotagliflozinu ve výši 400 mg. Další den byl podán opět sotagliflozin a spolu s ním kyselina mefenámová – první den v dávce 500 mg a od druhého do sedmého dne v dávkách 250 mg 4krát denně. Vlivem kyseliny mefenámové došlo ke zvýšení plochy pod křivkou sotagliflozinu o 52 % a jeho maximálních plazmatických koncentrací o 54 %. Ve druhé studii u zdravých dobrovolníků (Study INT14936) byl podáván rifampicin v opakovaných dávkách 600 mg jedenkrát denně, před zahájením podávání rifampicinu a spolu s jeho poslední dávkou byla podána jednorázová dávka sotagliflozinu ve výši 400 mg. Došlo ke snížení plochy pod křivkou sotagliflozinu o 60 % a ke snížení jeho maximálních plazmatických koncentrací o 40 %. Držitel rozhodnutí o registraci k tomu uvádí, že toto snížení expozice sotagliflozinu může snížit jeho účinnost. Pokud musí být současně se sotagliflozinem podáván induktor enzymů (např. rifampicin, fenytoin, fenobarbital, ritonavir), je třeba zvážit časté monitorování sérové koncentrace glukózy.
Závěr
Dapagliflozin lze považovat za nejcitlivější substrát UGT (UGT1A9). V případě dapagliflozinu tak lze očekávat klinicky relevantní lékové interakce s induktory UGT1A9 (např. rifampicinem nebo fenobarbitalem), případně i s inhibitory UGT1A9 (některé inhibitory tyrozinkinázy, např. nintedanib). U ostatních gliflozinů je jednak podíl produkovaných glukuronidů jako metabolitů menší a jednak se na jejich tvorbě podílí více izoenzymů UGT. Lze tedy důvodně očekávat menší riziko ovlivnění glukuronidace při inhibici, ale přibližně stejné riziko ovlivnění glukuronidace při indukci, protože známé induktory UGT mají polyfunkční efekt a indukují prakticky všechny izoenzymy UGT.
V současné době jsou inhibitory UGT1A9 používány v klinické praxi velmi omezeně. Nejsilnější inhibitory UGT1A9, tj. nesteroidní antiflogistika ze skupiny fenamátů, nejsou dokonce v ČR ani registrovány. S narůstajícím používáním inhibitorů tyrozinkinázy (např. dabrafenib, nintedanib, regorafenib, sorafenib nebo vandetanib), které mají in vitro silné inhibiční působení vůči UGT1A9, ale i dalším izoenzymům UGT, proto nelze v budoucnosti vyloučit „objev“ nových lékových interakcí.
Seznam použité literatury
- [1] van der Walt JS, Hong Y, Zhang L, et al. A Nonlinear Mixed Effects Pharmacokinetic Model for Dapagliflozin and Dapagliflozin 3‑O‑glucuronide in Renal or Hepatic Impairment. CPT Pharmacometrics Syst Pharmacol 2013; 2: e42.
- [2] Francke S, Mamidi RNVS, Solanki B, et al. In vitro metabolism of canagliflozin in human liver, kidney, intestine microsomes, and recombinant uridine diphosphate glucuronosyltransferases (UGT) and the effect of genetic variability of UGT enzymes on the pharmacokinetics of canagliflozin in humans. J Clin Pharmacol 2015; 55: 1061–1072.
- [3] Mori K, Saito R, Nakamaru Y, et al. Physiologically based pharmacokinetic‑pharmacodynamic modeling to predict concentrations and actions of sodium‑dependent glucose transporter 2 inhibitor canagliflozin in human intestines and renal tubules. Biopharm Drug Dispos 2016; 37: 491–506.
- [4] Lapham K, Callegari E, Cianfrogna J, et al. In vitro charakterization of ertugliflozin glucuronidation. Abstracts from the 22nd North American ISSX Meetion, July 15–19, 2018, Montreal, Canada, 48 (abstrakt P16). Dostupné na: https://bib.irb.hr/datoteka/947954.22nd_north_american_issx_meeting_abstract_book.pdf
- [5] Chen LZ, Jungnik A, Mao Y, et al. Biotransformation and mass balance of the SGLT2 inhibitor empagliflozin in healthy volunteers. Xenobiotica 2015; 45: 520–529.
- [6] SPC Forxiga® (dapagliflozin), 3/2021. Dostupné na: https://www.ema.europa.eu/en/documents/product‑information/forxiga‑epar‑product‑information_cs.pdf
- [7] SPC Invokana® (kanagliflozin), 1/2020. Dostupné na: https://www.ema.europa.eu/en/documents/product‑information/invokana‑eparproduct‑information_cs.pdf
- [8] SPC Jardiance® (empagliflozin), 7/2021. Dostupné na: https://www.ema.europa.eu/en/documents/product‑information/jardiance‑eparproduct‑information_cs.pdf
- [9] SPC Steglatro® (ertugliflozin), 8/2020. Dostupné na: https://www.ema.europa.eu/documents/product‑information/steglatro‑epar‑productinformation_cs.pdf
- [10] SPC Zynquista® (sotagliflozin), 10/2020. Dostupné na: https://www.ema.europa.eu/en/documents/product‑information/zynquista‑eparproduct‑information_cs.pdf
- [11] EMA CHMP. Assessment Report. Zynquista® (sotagliflozin), sanofi‑aventis, 2/2019. Dostupné na: https://www.ema.europa.eu/en/documents/assessment‑report/zynquista‑epar‑public‑assessment‑report_en.pdf
- [12] FDA Briefing Document: Endocrinologic & Metabolic Drugs Advisory Committee. SAR439954 – Sotagliflozin, 12/2018. Dostupné na: https://www.fda.gov/media/121285/download
- [13] Kasichayanula S, Liu X, Pe Benito M, et al. The influence of kidney function on dapagliflozin exposure, metabolism and pharmacodynamics in healthy subjects and in patients with type 2 diabetes mellitus. Br J Clin Pharmacol 2013; 76: 432–444.
- [14] Lv X, Xia Y, Finel M, et al. Recent progress and challenges in screening and characterization of UGT1A1 inhibitors. Acta Pharm Sin B 2019; 9: 258–278.
- [15] Kasichayanula S, Lacreta F, Griffen SC. Effects of rifampin and mefenamic acid on the pharmacokinetics and pharmacodynamics of dapagliflozin. Diabetes Obes Metab 2013; 15: 280–283.
- [16] Naagaard MD, Chang R, Någård M, et al. Common UGT1A9 polymorphisms do not have a clinically meaningful impact on the apparent oral clearance of dapagliflozin in type 2 diabetes mellitus. Br J Clin Pharmacol 2022; 88: 1942–1946.
- [17] Hoeben E, de Winter W, Neyens M, et al. Population Pharmacokinetic Modeling of Canagliflozin in Healthy Volunteers and Patients with Type 2 Diabetes Mellitus. Clin Pharmacokinet 2016; 55: 209–223.
- [18] Pattanawongsa A, Chau N, Rowland A, Miners JO. Inhibition of Human UDP‑Glucuronosyltransferase Enzymes by Canagliflozin and Dapagliflozin: Implications for Drug‑Drug Interactions. Drug Metab Dispos 2015; 43: 1468–1476.
- [19] Devineni D, Vaccaro N, Murphy J, et al. Effects of rifampin, cyclosporine A, and probenecid on the pharmacokinetic profile of canagliflozin, a sodium glucose co‑transporter 2 inhibitor, in healthy participants. Int J Clin Pharmacol Ther 2015; 53: 115–128.
- [20] FDA Center for Drug Evaluation and Research. Clinical Pharmacology and Biopharmaceutic Review. Application Number 204629Orig1s000. Jardiance® (empagliflozin), Boehringer Ingelheim Pharmaceuticals, 6/2014. Dostupné na: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/204629Orig1s000ClinPharmR.pdf
- [21] Macha S, Koenen R, Sennewald R, et al. Effect of gemfibrozil, rifampicin, or probenecid on the pharmacokinetics of the SGLT2 inhibitor empagliflozin in healthy volunteers. Clin Ther 2014; 36: 280–290.
- [22] Callegari E, Lin J, Tse S, et al. Physiologically‑Based Pharmacokinetic Modeling of the Drug‑Drug Interaction of the UGT Substrate Ertugliflozin Following Co‑Administration with the UGT Inhibitor Mefenamic Acid. CPT Pharmacometrics Syst Pharmacol 2021; 10: 127–136.
- [23] Dawra VK, Sahasrabudhe V, Liang Y, et al.. Effect of Rifampin on the Pharmacokinetics of Ertugliflozin in Healthy Subjects. Clin Ther 2018; 40: 1538–1554.