Inovativní lékové formy pro těžce rozpustná léčiva
V současné farmakoterapii neustále přibývá léčivých látek, které jsou velmi těžce rozpustné ve vodě, což také často limituje i jejich systémovou absorpci po perorálním podání. Odhaduje se, že do této kategorie spadá až 40 % dnes běžně používaných léčiv. Tyto látky jsou však obvykle dobře rozpustné v tucích, přesněji v lipofilních rozpouštědlech. Doposud se jejich špatná rozpustnost ve vodě řešila především úpravou léčiva nebo lékové formy. Teprve nedávno se objevil nový trend využít dobrou rozpustnost léčiv v lipofilních rozpouštědlech, popřípadě použít přímo kapalná lipofilní léčiva. Lipofilní kapalina s eventuální přísadou tenzidů a kosolventů se pak rovnou plní nejčastěji do měkkých nebo tvrdých tobolek, vznikají tzv. samoemulgující systémy. Další možností je navázat léčivo v kapalné fázi na vysoce porézní sorbent za vzniku práškové směsi, kterou je možné lisovat do tablet nebo plnit do tvrdých tobolek – tzv. systémy kapalina v pevné fázi. Oba systémy představují směr, kterým se v současnosti začala ubírat řada výrobců. Oba systémy mají tu výhodu, že obsahují již rozpuštěné léčivo a tím zvyšují jeho vstřebávání, respektive biodostupnost. Přestože na trhu již existuje řada samoemulgujících systémů, systémy kapalina v pevné fázi jsou zatím ve stadiu klinického výzkumu.
Úvod
Rozpustnost léčiva ve vodě patří mezi nejdůležitější parametry ovlivňující dosažení jeho požadované koncentrace v systémové cirkulaci, která je schopna v organismu vyvolat zamýšlenou farmakologickou odpověď. Dle platného Českého lékopisu je z fyzikálně-chemického hlediska možné léčiva rozdělit na základě jejich rozpustnosti do sedmi skupin (tab. 1). Toto členění je však z farmakologického hlediska obvykle nedostačující, neboť zde není zohledněna terapeutická dávka léčiva. Jinými slovy, nezáleží na tom, jak je léčivo rozpustné ve vodě, ale zda se podaná dávka po jeho požití rozpustí. V případě některých látek je totiž možné získat farmakologickou odpověď již po 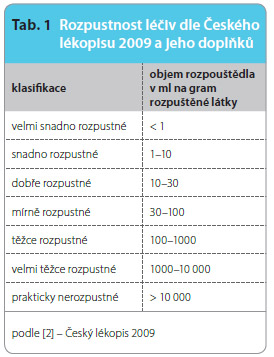 podání velmi malého množství léčiva (např. hormony), zatímco u jiných (antibiotika apod.) je podání vyšší dávky nezbytné. Z toho vyplývá, že i malé množství špatně rozpustného léčiva se může v gastrointestinálním traktu (GIT) zcela rozpustit, zatímco vyšší množství relativně dobře rozpustné léčivé látky se zde naopak rozpustit nemusí [1].
podání velmi malého množství léčiva (např. hormony), zatímco u jiných (antibiotika apod.) je podání vyšší dávky nezbytné. Z toho vyplývá, že i malé množství špatně rozpustného léčiva se může v gastrointestinálním traktu (GIT) zcela rozpustit, zatímco vyšší množství relativně dobře rozpustné léčivé látky se zde naopak rozpustit nemusí [1].
Z tohoto důvodu byl v devadesátých letech minulého století navržen biofarmaceutický klasifikační systém (BCS), který rozděluje léčivé látky do čtyř tříd na základě jejich rozpustnosti ve vodném prostředí a na základě gastrointestinální prostupnosti – permea-
bility (obr. 1). Za vysoce rozpustná léčiva se v rámci BCS považují ta, jejichž nejvyšší dávka se rozpustí ve 250 ml vodného pufru obvykle v rozmezí pH 1–8, zatímco jako vysoce prostupné se označují látky, které mají u člověka míru absorpce z GIT nejméně 90 % z podané dávky [3]. Uvádí se, že až 40 % běžně používaných léčiv a až 70 % nově syntetizovaných látek vykazuje nízkou rozpustnost ve vodě a je možné je zařadit je do BCS třídy II (nízká rozpustnost, vysoká prostupnost) nebo IV (nízká rozpustnost,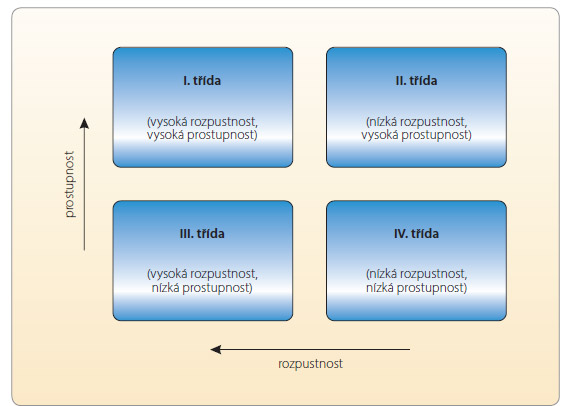 nízká prostupnost) [4]. Špatná rozpustnost je obvykle spjata s nízkou biologickou dostupností, proto léčivé látky řadící se do těchto dvou tříd způsobují řadu problémů během formulace pevných lékových forem určených pro systémovou absorpci léčiva. Zvyšování biologické dostupnosti látek špatně rozpustných ve vodě se tudíž stalo jedním z trendů moderní farmaceutické technologie a v odborné literatuře bylo popsáno několik postupů vedoucích ke zvýšení jejich rozpustnosti, respektive biologické dostupnosti. Mezi tyto metody je možné mimo jiné zařadit např. mikronizaci [5] (např. Novofem® obsahující mikronizovaný 17β-estradiol), přípravu nanokrystalů [6] (např. Rapamune® s obsahem sirolimu), formulaci pevných disperzí [7] (např. Prograf® obsahující takrolimus) nebo zapracování těžce rozpustného léčiva do pevné lékové formy v kapalné formě za pomoci samoemulgujících systémů či systémů kapalina v pevné fázi.
nízká prostupnost) [4]. Špatná rozpustnost je obvykle spjata s nízkou biologickou dostupností, proto léčivé látky řadící se do těchto dvou tříd způsobují řadu problémů během formulace pevných lékových forem určených pro systémovou absorpci léčiva. Zvyšování biologické dostupnosti látek špatně rozpustných ve vodě se tudíž stalo jedním z trendů moderní farmaceutické technologie a v odborné literatuře bylo popsáno několik postupů vedoucích ke zvýšení jejich rozpustnosti, respektive biologické dostupnosti. Mezi tyto metody je možné mimo jiné zařadit např. mikronizaci [5] (např. Novofem® obsahující mikronizovaný 17β-estradiol), přípravu nanokrystalů [6] (např. Rapamune® s obsahem sirolimu), formulaci pevných disperzí [7] (např. Prograf® obsahující takrolimus) nebo zapracování těžce rozpustného léčiva do pevné lékové formy v kapalné formě za pomoci samoemulgujících systémů či systémů kapalina v pevné fázi.
Samoemulgující systémy
Samoemulgující systémy (self-emulsifying drug delivery systems, SEDDS) se řadí mezi tzv. lipofilní formulace a jsou definovány jako izotropní směsi léčiva, olejů, povrchově aktivních látek a v některých případech také hydrofilních kosolventů a dalších emulgátorů. Jejich hlavním specifikem je schopnost tvořit po podání do gastrointestinálního traktu, respektive po mírném promísení s vodnou fází, jemné emulze (popř. mikroemulze nebo nanoemulze) olej ve vodě (o/v) [8]. Na základě velikosti kapek vnitřní fáze vzniklé emulze je možné samoemulgující formulace rozdělit na samoemulgující systémy (250 nm – 5 µm), samomikroemulgující systémy (100–250 nm) a samonanoemulgující systémy (< 100 nm) [9]. Díky velkému povrchu vzniklých kapének se léčivo snadněji uvolňuje do intestinálních tekutin, čímž dochází k jeho rychlejší absorpci do systémového oběhu, a tím i ke zvýšení jeho biologické dostupnosti, což je hlavní výhodou těchto formulací [10]. K dalším výhodám pak patří snížení vlivu potravy na rychlost a množstv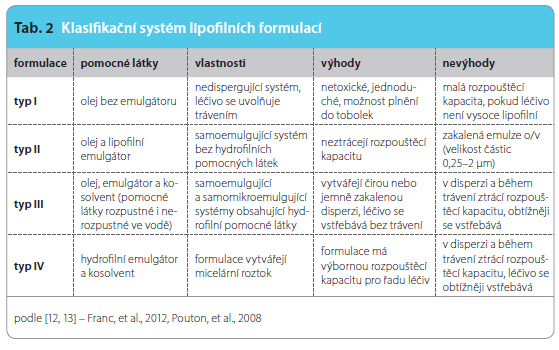 í uvolněného léčiva, konstantní rychlost vstřebávání léčivé látky a případně i možnost formulace samoemulgujících lékových forem s řízeným uvolňováním [11].
í uvolněného léčiva, konstantní rychlost vstřebávání léčivé látky a případně i možnost formulace samoemulgujících lékových forem s řízeným uvolňováním [11].
Lipofilní formulace dělíme do čtyř skupin (tab. 2), které se liší zejména přítomností emulgátorů a kosolventů. Do první skupiny patří léky tvořené pouze oleji, nejčastěji ve formě acylglycerolů, s rozpuštěnými či přimísenými léčivy. Léčiva se zde do organismu uvolňují obvykle až po natrávení oleje trávicími enzymy. Do druhé skupiny se řadí oleje s obsahem lipofilních emulgátorů a léčiv. Emulgátory pak vedle procesu natrávení pomáhají i k uvolnění léčiva do trávicích šťáv. Třetí skupinu tvoří léky obsahující vedle emulgátou i hydrofilní kosolventy. Jejich výhodou je, že uvolňují léčiva bez natrávení.
Poslední a čtvrtou skupinou jsou léky neobsahující olej, ale pouze hydrofilní kosolvent s emulgátorem, kde je léčivo přítomno ve formě micelárního roztoku. Jejich nevýhodou je, že při styku s trávicími šťávami občas vytvářejí viskózní ložiska, která zabraňují rychlému vstřebávání [12].Samoemulgující formulace jsou obvykle připravovány jako kapaliny, které se následně plní do měkkých a tvrdých želatinových tobolek. Tato podoba SEDDS má však řadu nevýhod, jako jsou např. vyšší výrobní náklady, nízká stabilita, možné inkompatibility mezi složkami SEDDS a pomocnými látkami obalu a omezené možnosti podoby finální lékové formy [14]. Některé z těchto nevýhod mohou být odstraněny převedením SEDDS do pevné podoby (pevné samoemulgující systémy) pomocí extruze/sféronizace, vlhké granulace, sprejového sušení či techniky přípravy systémů kapalina v pevné fázi [15].
Příkladem samoemulgujících systémů dostupných na českém trhu mohou být Sandimmun® (cyklosporin A), Vesanoid® (tretinoin), Aptivus® (tipranavir) a samomikroemulgující systém Sandimmun Neoral® (cyklosporin A). V zahraničí jsou pak navíc dostupné přípravky Accutane® (isotretinoin), Panimum Bioral® (cyklosporin), Gengraf® (cyklosporin A), Kaletra® (kombinace lopinaviru a ritonaviru), Fortovase® (sanquinavir) a Agenerase® (amprenavir).
Systémy kapalina v pevné fázi
Systémy kapalina v pevné fázi, někdy také označované na základě jejich anglického názvu jako liquisolid systémy (LSS), jsou moderní formulace schopné zvyšovat biologickou dostupnost špatně rozpustných léčiv. Z historického hlediska se tyto systémy vyvinuly z tzv. práškových roztoků, které se získávaly inkorporací roztoku léčiva v netěkavém rozpouštědle do struktury inertního nosiče s velkým povrchem částic, jako je např. oxid křemičitý. Tyto přípravky však nebylo možné transformovat do jiné než práškové podoby, jelikož jejich vlastnosti neumožňovaly lisování do tablet. I přes snahu upravit lisovatelnost těchto systémů přidáním plniv zlepšujících stlačitelnost (např. mikrokrystalická celulosa) nebyly vlastnosti přípravků nikdy přizpůsobeny požadavkům farmaceutického průmyslu [16, 17].
LSS jsou tedy technologickým vylepšením práškových roztoků a hlavním principem jejich přípravy (obr. 2) je nasorbování léčiva v kapalné fázi (roztok, suspenze, emulze nebo výše zmíněný samoemulgující systém) na vysoce porézní nosič, který je následně obalen velmi jemným materiálem s vysokým povrchem částic (obalovací materiál), za vzniku suchého nepřilnavého prášku s vlastnostmi vhodnými pro další zpracování (plnění do tobolek, lisování do tablet apod.) [16].
Oproti běžně používanému zapracování kapalných léčiv do pevné lékové formy (příprava želatinových perel) mají LSS řadu výhod, mezi které lze zařadit: jednoduchost zpracování, nízké výrobní náklady, minimalizaci vlivu pH na rychlost uvolňování léčiva, zlepšení 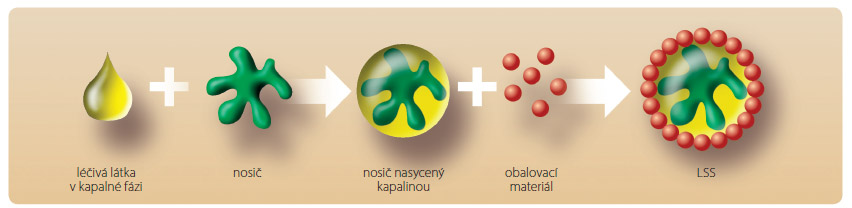 disolučního profilu, zvýšení biologické dostupnosti těžce rozpustných léčivých látek a možnost přípravy lékové formy s řízeným uvolňováním léčiv dobře rozpustných ve vodě [18].
disolučního profilu, zvýšení biologické dostupnosti těžce rozpustných léčivých látek a možnost přípravy lékové formy s řízeným uvolňováním léčiv dobře rozpustných ve vodě [18].
Na zvyšování biologické dostupnosti léčiv formulovaných do podoby LSS se podílí několik mechanismů. Prvním z nich je přítomnost disperze léčiva na celém povrchu nosiče, díky čemuž se léčivo snadněji uvolní z lékové formy. Navíc je díky přítomnosti hydrofilního rozpouštědla, které je nezbytné pro převedení účinné látky do kapalné podoby, zvýšena smáčivost přípravku disolučním médiem. Avšak hlavním principem zvyšování biologické dostupnosti pomocí těchto systémů je přítomnost léčiva v kapalné podobě, jež se po podání do GIT již nemusí rozpouštět, a léčivo je tedy ihned dostupné pro absorpci do systémového oběhu [19].
V současné době se na českém ani zahraničním trhu nenachází žádný komerčně dostupný systém kapalina v pevné fázi. V odborné literatuře však byla popsána řada liquisolid systémů se zapracovaným kapalným léčivem (klofibrát [20]), roztokem nebo suspenzí špatně rozpustného léčiva ve vybraném hydrofilním rozpouštědle (atorvastatin [21], griseofulvin [22], karbamazepin [23], indometacin [24] apod.) či s kapalným samoemulgujícím systémem (cyklosporin A [25]). Navíc byly touto technikou připraveny i orálně dispergovatelné tablety s obsahem aceklofenaku [26] a tablety s řízeným uvolňováním léčiv dobře rozpustných ve vodě (propranolol [27], tramadol [28], theofylin [29] aj.).
Závěr
Zapracování špatně rozpustného léčiva v kapalné podobě do pevné lékové formy představuje nový přístup ke zvyšování biologické dostupnosti těchto látek. Samoemulgující systémy spolu se systémy kapalina v pevné fázi představují novou strategii při vývoji pevných perorálních lékových forem obsahujících špatně rozpustná léčiva. Díky jeho přítomnosti v již rozpuštěné podobě je v gastrointestinálním traktu vynechán nejvíce limitující krok absorpce do systémového oběhu, a to rozpouštění léčivé látky. Zatímco samoemulgující systémy je možné najít i mezi komerčně dostupnými přípravky, systémy kapalina v pevné fázi jsou v samotném počátku svého vývoje a očekává se od nich, že budou hrát důležitou roli při přípravě moderních pevných lékových forem.
Seznam použité literatury
- [1] Okáčová L, Vetchý D, Franc A, et al. Zvýšení biodostupnosti těžce rozpustných léčivých látek jejich modifikací. Chem Listy 2010; 104: 21–26.
- [2] Český lékopis 2009 1. díl. Praha, Grada Publishing 2009.
- [3] Yazdanian M, Briggs K, Jankovsky C, Hawi A. The „High Solubility“ Definition of the Current FDA Guidance on Biopharmaceutical Classification System May Be Too Strict for Acidic Drugs. Pharm Res 2004; 21: 293–299.
- [4] Kawabata Y, Wada K, Nakatani M, et al. Formulation design for poorly water-soluble drugs bases on biopharmaceutics classifiation system: Basic approaches and practical application. Int J Pharm 2011; 420: 1–10.
- [5] Rasenack N, Müller BW. Dissolution Rate Enhancement by in Situ Micronization of Poorly Water-Soluble Drugs. Pharm Res 2002; 19: 1894–1900.
- [6] Keck CM, Müller RH. Drug nanocrystals of poorly soluble drugs produced by high pressure homogenization. Eur J Pharm Biopharm 2006; 62: 3–16.
- [7] Vasconcelos T, Sarmento B, Costa P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov Today 2007; 12: 1068–1075.
- [8] Gursoy RN, Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother 2004; 58: 173–182.
- [9] Kohli K, Chopra S, Dhar D, et al. Self-emulsifying drug delivery systems: An approach to enhance oral bioavailability. Drug Discov Today 2010; 15: 958–965.
- [10] Kommuru TR, Gurley B, Khan MA, Reddy IK. Self-emulsifying drug delivery systems (SEDDS) of coenzyme Q10: formulation development and bioavailability assessment. Int J Pharm 2001; 212: 233–246.
- [11] Kumar A, Sharma S, Kamble R. Self emulsifying drug delivery system (SEEDS): Future aspects. Int J Pharm Pharm Sci 2010; 2: 7–13.
- [12] Franc A, Vetchý D, Smilková L, et al. Lipofilní formulace pro zvýšení biodostupnosti těžce rozpustných léčivých látek. Chem Listy 2012; 106: 3–9.
- [13] Pouton CW, Porter CJ. Formulation of lipid-based delivery systems for oral administration: materials, methods and strategies. Adv Drug Deliv Rev 2008; 60: 625–637.
- [14] Kallakunta VR, Bandari S, Jukanti R, Veerareddy PR. Oral self emulsifying powder of lercanidipine hydrochloride: Formulation and evaluation. Powder Technol 2012; 221: 375–382.
- [15] Tang B, Cheng G, Gu J, Xu C. Development of solid self-emulsifying drug delivery systems: preparation techniques and dosage forms. Drug Discov Today 2008; 13: 606–612.
- [16] Kavitha K, Lova Raju KNS, et al. Effect of dissolution rate by liquisolid compacts approach: An Overview. Der Pharmacia Lettre 2011; 3: 71–83.
- [17] Kulkarni AS, Aloorkar NH, Mane MS, Gaja JB. Liquisolid Systems: A Review. Int J Pharm Sci Nanotech 2010; 3: 795–802.
- [18] Vraníková B, Gajdziok J. Liquisolid systems and aspects influencing their research and development. Acta Pharm 2013; 63: 447–465.
- [19] Nokhodchi A, Hentzschel CM, Leopold CS. Drug release from liquisolid systems: speed it up, slow it down. Expert Opin Drug Deliv 2011; 8: 191–205.
- [20] Nagabandi VK, Ramarao T, Jayaveera KN. Liquisolid Compacts: A Novel Approach to Enhance Bioavailability of Poorly Soluble Drugs. Int J Pharm Bio Sci 2011; 1: 89–102.
- [21] Gubbi SR, Jarag R. Formulation and characterization of atorvastatin calcium liquisolid compacts. Asian J Pharm Sci 2010; 5: 50–60.
- [22] Hentzschel CM, Alnaief M, Smirnova I, et al. Enhancement of griseofulvin release from liquisolid compacts. Eur J Pharm Biopharm 2011; 80: 130–135.
- [23] Tayel SA, Soliman II, Louis D. Improvement of dissolution properties of Carbamazepine through application of the liquisolid tablet technique. Eur J Pharm Biopharm 2008; 69: 342–347.
- [24] Javadzadeh Y, Siahi MR, Asnaashari S, Nokhodchi A. Liquisolid technique as a tool for enhancement of poorly water-soluble drugs and evaluation of their physicochemical properties. Acta Pharm 2007; 57: 99–109.
- [25] Zhao X, Zhou YQ, Potharaju S, et al. Development of a self micro-emulsifying tablet of cyclosporine-A by the liquisolid compact technique. Int J Pharm Sci Res 2011; 2: 2299–2308.
- [26] Yadav AV, Shete AS, Dabke AP. Formulation and Evaluation of Orodispersible Liquisolid Compacts of Aceclofenac. Indian J Pharm Educ Res 2010; 44: 227–235.
- [27] Javadzadeh Y, Musaalrezaei L, Nokhodchi A. Liquisolid technique as a new approach to sustain propranolol hydrochloride release from tablet matrices. Int J Pharm 2008; 362: 102–108.
- [28] Gonjari ID, Karmarkar AB, Hosmani AH. Evaluation of in vitro dissolution profile comparison methods of sustained release tramadol hydrochloride liquisolid compact formulations with marketed sustained release tablets. Dig J Nanomater Bios 2009; 4: 651–661.
- [29] Nokhodchi A, Aliakbar R, Desai S, Javadzadeh Y. Liquisolid compacts: The effect of cosolvent and HPMC in theophylline release. Colloid Surface B 2010; 79: 262–269.