Lékové interakce antimigrenik (1. část)
Souhrn:
Suchopár J, Suchopár Š, Prokeš M. Lékové interakce antimigrenik (1. část). Remedia 2021; 31: 284–291.
Antimigrenika jsou léčiva, která, až na výjimky, mají obecně poměrně značný potenciál k lékovým interakcím. Klasická antimigrenika působí serotonergně a vzácně mohou přispívat ke vzniku serotoninového syndromu, zejména u pacientů již léčených jinými serotonergními léky nebo užívajícími tramadol jako analgetikum. Metabolismus některých z nich je závislý na CYP3A4 nebo MAO‑A, jejichž inhibice může v některých případech vést ke klinicky významnému zvýšení expozice a vzniku nežádoucích účinků nebo toxicity. Nová antimigrenika ze skupiny tzv. gepantů představují v oblasti lékových interakcí jistou výzvu, neboť tyto léky mají poměrně značný počet lékových interakcí, které jsou klinicky významné a vyžadují úpravy dávek nebo intervalu mezi dávkami, resp. četnosti dávek. Biologická léčiva naopak představují bezpečnou alternativu terapie, neboť lékové interakce nemají a je vysoce nepravděpodobné, že by se v budoucnosti na tomto konstatování cokoliv změnilo.
Summary:
Suchopar J, Suchopar S, Prokes M. Drug interactions of antimigraine drugs (part 1). Remedia 2021; 31: 284–291.
Antimigraine drugs are drugs that, with a few exceptions, generally have a relatively high potential for drug interactions. Classical antimigraine drugs have a serotonergic effect and may rarely contribute to serotonin syndrome, especially in patients already treated with other serotonergic drugs such as most antidepressants or taking tramadol as an analgesic. The metabolism of some of them is dependent on CYP3A4 or MAO‑A, the inhibition of which may in some cases lead to a clinically significant increase in exposure and the development of adverse effects or toxicity. New antimigraine drugs from the group of so‑called gepants represent a certain challenge in the field of drug interactions, as these drugs have a high number of drug interactions that are clinically significant and require adjustments of dose, dose interval, or dose frequency. Biological drugs, on the other hand, are a safe alternative to therapy because they do not possess drug‑interaction activity and it is highly unlikely that anything will change in the future.
Key words: drug interactions, antimigraine drugs, ergotamine, dihydroergotamine, triptans, lasmiditan, rimegepant, ubrogepant, fremanezumab, erenumab, galcanezumab
Úvodní informace
Terapie a profylaxe migrény se za poslední tři roky výrazně změnily. Do terapeutické palety antimigrenik se zařadily čtyři nové lékové skupiny s novým mechanismem účinku. Tyto léky mají odlišné mechanismy působení, velmi dobrou účinnost a nižší výskyt nežádoucích účinků, kterými se vyznačují klasická antimigrenika. Část z těchto léků však má klinicky významné lékové interakce. Předmětem tohoto článku jsou právě lékové interakce antimigrenik, a to jak klasických, tak i těch nových.
Celosvětový náskok v uvádění nových léčiv mají ve Spojených státech amerických, antimigrenika z toho nevyjímaje. K první registraci nových antimigrenik zde došlo v první polovině roku 2018, když 17. května 2018 americký Úřad pro kontrolu potravin a léčiv (FDA) schválil erenumab (Aimovig) jako první monoklonální protilátku proti receptoru CGRP (calcitonin gene related peptide). CGRP je neuropeptid obsahující 37 aminokyselin, který je odvozen od kalcitoninu a produkován v periferních a centrálních neuronech; fyziologicky se uplatňuje v přenosu nocicepce a v kardiovaskulární homeostáze (jako mohutné vazodilatans a látka s pozitivním chromotropním účinkem). Následně byly schváleny další biologické léky určené k profylaxi migrény, a to galkanezumab (Emgality) v září 2018, fremanezumab (Ajovy) v září 2019 a s malým odstupem pak eptinezumab (Vyepti) v únoru 2020, všechny tři tyto monoklonální protilátky mají odlišný mechanismus účinku, neboť se váží přímo na CGRP. Dalším pokrokem v paletě nových antimigrenik bylo schválení použití perorálních receptorových antagonistů CGRP, a to ubrogepantu (Ubrelvy), který FDA schválil 23. prosince 2019, a dále rimegepantu (Nurtec ODT) s datem schválení 27. února 2020. Jako zatím poslední léčivo ze skupiny nových antimigrenik schválil FDA v říjnu 2019 prvního zástupce selektivních agonistů serotoninového 5 HT1F receptoru lasmiditan (Reyvow).
K první změně v Evropě došlo 26. července 2018, kdy Evropská léková agentura (EMA) schválila erenumab (Aimovig). Po jisté latenci následovalo schválení fremanezumabu (Ajovy) v březnu 2019 a galkanezumabu (Emgality) v listopadu 2019. Do systému úhrad v České republice jako první vstoupil erenumab (Aimovig), a to 1. února 2020, následovaný fremanezumabem (Ajovy) v květnu 2020 a galkanezumabem (Emgality) v říjnu 2020. Ubrogepant, rimegepant, lasmiditan ani eptinezumab zatím nejsou v Evropě registrovány.
Aktuálně lze antimigrenika rozdělit následovně:
- Klasická antimigrenika
námelové alkaloidy: ergotamin, dihydroergotamin, kodergokrin (dříve dihydroergotoxin)
triptany: sumatriptan, naratriptan, zolmitriptan, rizatriptan, almotriptan, eletriptan, frovatriptan
- Nové skupiny antimigrenik
ditany: lasmiditan
gepanty: rimegepant, ubrogepant
monoklonální protilátky proti CGRP: eptinezumab, fremanezumab, galkanezumab
monoklonální protilátky proti receptoru CGRP: erenumab
V tomto přehledu se nebudeme zabývat léčivy určenými k tlumení bolesti (paracetamol, nesteroidní antirevmatika, metamizol, kodein), neuvádíme také léčiva používaná k profylaxi migrény ze skupiny antiepileptik (např. topiramát, kyselinu valproovou) ani blokátorů kalciového kanálu (např. cinarizin nebo verapamil) či betablokátorů (např. atenolol nebo metoprolol). Tato léčiva však mají značné množství lékových interakcí, z nichž některé byly popsány teprve nedávno. Část z lékových interakcí těchto lékových skupin je klinicky významných, a je proto třeba při terapii migrény na ně myslet.
Lékové interakce námelových
alkaloidů
Námelové alkaloidy byly v minulosti používány při terapii i profylaxi migrény poměrně často, což bylo dáno zejména nedostupností jiné účinné terapie. Také v současnosti se s jejich použitím lze setkat, např. pokud jsou předepsány magistraliter (v lékové formě čípků atd.). Všechny námelové alkaloidy, které se při terapii migrény používaly nebo stále používají, jsou citlivé substráty cytochromu P450 3A4 (CYP3A4). Touto cestou se metabolizuje významně více než 50 % podané dávky. Inhibice CYP3A4 je tak hlavním mechanismem lékových interakcí námelových alkaloidů a tento typ lékových interakcí pravděpodobně přispěl k omezení používání námelových alkaloidů, které doporučil Výbor pro humánní léčivé přípravky EMA [1]. Od roku 2013 tak nelze používat námelové alkaloidy při profylaxi migrény a jejich podání je možné výhradně při zvládání migrenózní akutní ataky. Aktuálně je v ČR dostupný pouze kodergokrin, který je obsažen v léčivém přípravku Secatoxin.
Na základě studií in vitro a in vivo se má za prokázané, že při souběžném perorálním podávání ergotaminu nebo dihydroergotaminu s inhibitory CYP3A4 dojde ke zvýšení jejich plazmatických koncentrací [2]. Stejné poznání platí též pro kodergokrin [3]. Důsledky zvýšení expozice jednotlivým námelovým alkaloidům nejsou zcela shodné. Klinicky významně zvýšené koncentrace ergotaminu nebo dihydroergotaminu způsobují ergotismus, jehož podstatou je vazokonstrikce. Manifestuje se nejčastěji jako vazospasmus v končetinách (většinou dolních, někdy i horních, zřídka jen horních) s chladnými akrálními částmi a paresteziemi, déletrvající porucha prokrvení může vyústit v gangrénu s nutnou amputací. Může se však projevit jako cerebrální nebo koronární vazospasmus nebo také střevní ischemie. Klinicky významně zvýšené koncentrace kodergokrinu k ergotismu nevedou, způsobují však hypotenzi a bradykardii, přičemž hypotenze může být klinicky vysoce závažná a může přecházet v komatózní stav a vést k srdeční zástavě.
Bylo popsáno mnoho desítek kazuistik klinicky vysoce závažných lékových interakcí ergotaminu nebo dihydroergotaminu s inhibitory CYP3A4, a to jak se silnými, kde by se lékové interakce daly očekávat, tak i se středně silnými, nebo dokonce slabými inhibitory CYP3A4. Databáze nežádoucích účinků FDA (FDA Adverse Event Reporting System, FAERS) uvádí k 31. 12. 2020 celkem 786 závažných nežádoucích účinků v důsledku podávání ergotaminu a dihydroergotaminu, z toho 43 s fatálním zakončením. Celkem je v databázi FAERS popsáno 69 případů lékových interakcí ergotaminu nebo dihydroergotaminu s inhibitory CYP3A4, z toho tři s fatálním zakončením [4].
Přehled vybraných kazuistik ergotismu
v důsledku souběžného podávání ergotaminu nebo
dihydroergotaminu s inhibitory CYP3A4 uvádíme v tabulce 1.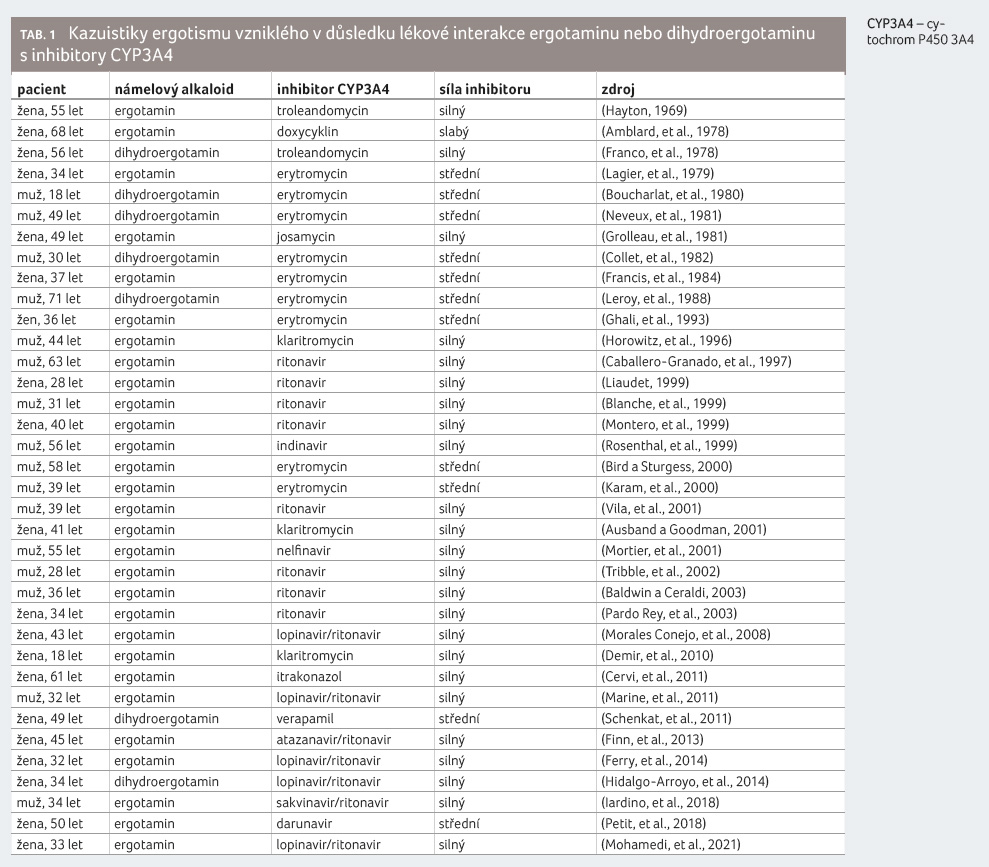
Pozoruhodné je, že ačkoliv první kazuistika byla popsána již v roce 1969 a cílená farmakokinetická studie se uskutečnila v roce 1991, jsou kazuistiky ergotismu, včetně závažných forem s fatálním zakončením, popisovány do současnosti, zatím poslední v roce 2021. Dosud nebyla popsána žádná kazuistika klinicky významné lékové interakce v případě kodergokrinu, což je jednak dáno jeho převažujícím vazodilatačním efektem v důsledku α sympatolytického účinku tohoto námelového alkaloidu a jednak také tím, že je při terapii migrény používán významně méně často.
Kodergokrin, který byl v minulosti označován jako dihydroergotoxin, je směsí tří dihydrogenovaných námelových alkaloidů (dihydroergokristinu, dihydroergokorninu a α a β dihydroergokryptinu). Jak již bylo uvedeno, kodergokrin představuje vysoce citlivý substrát CYP3A4. Souběžné podávání jedné z jeho složek – α dihydroergokryptinu – se středně silným inhibitorem CYP3A4 erytromycinem vedlo u zdravých dobrovolníků k excesivnímu zvýšení expozice α dihydroergokryptinu na 16,5násobek (8,7−31,5násobek na 95% hladině spolehlivosti) a k jen o něco menšímu zvýšení jeho maximálních plazmatických koncentrací, na 9,5násobek (6,5−13,9násobek na 95% hladině spolehlivosti) [5].
Výrazný vliv souběžně podávaného inhibitoru CYP3A4 na farmakokinetické vlastnosti perorálně podaného dihydroergotaminu byl zaznamenán v klinické studii provedené již 10 let před studií s kodergokrinem, a to s použitím středně silného inhibitoru CYP3A4 miokamycinu (makrolidové antibiotikum s podobným inhibičním účinkem vůči CYP3A4, jako má erytromycin, a slabším inhibičním účinkem, než má registrovaný klaritromycin). V uvedené studii u zdravých dobrovolníků byl po dobu devíti dnů perorálně podáván miokamycin v dávkách 800 mg dvakrát denně, přičemž před zahájením podávání miokamycinu a v osmém dni jeho užívání byla perorálně podána jednorázová dávka dihydroergotaminu ve výši 9 mg [2]. Vlivem miokamycinu došlo k 3−40násobnému zvýšení maximálních plazmatických koncentrací dihydroergotaminu (v průměru došlo ke zvýšení o 2 246 %). Změna velikosti expozice dihydroergotaminu nebyla ve výsledcích studie uvedena.
Na druhou stranu bylo v další studii zjištěno, že při nazální aplikaci dihydroergotaminu spolu se souběžným podáváním silného inhibitoru CYP3A4 ketokonazolu nebyly zjištěny klinicky významné změny farmakokinetických vlastností dihydroergotaminu. V této studii u zdravých dobrovolníků byl perorálně podáván ketokonazol v dávkách 400 mg jednou denně po dobu čtyř dnů, před zahájením podávání ketokonazolu a za jednu hodinu po jeho poslední dávce byla inhalačně aplikována jednorázová dávka dihydroergotaminu ve výši 1 mg [6]. Vlivem ketokonazolu došlo ke zvýšení plochy pod křivkou dihydroergotaminu v průměru o 18 % (od snížení o 4 % po zvýšení o 44 % na 90% hladině spolehlivosti), avšak tento rozdíl nebyl statisticky významný. Zdá se tedy, že léková interakce dihydroergotaminu s inhibitory CYP3A4 je klinicky významná jen při perorálním podání.
V každém případě platí, že souběžné perorální podávání námelových alkaloidů se silnými a středně silnými inhibitory CYP3A4 je v případě ergotaminu nebo dihydroergotaminu z důvodu rizika vzniku kritické ischemie kontraindikováno a v případě kodergokrinu je z důvodu závažné hypotenze až komatózního stavu a bradykardie nezbytné se mu vyhnout.
Zda je z hlediska lékových interakcí bezpečná rektální, nazální, nebo injekční aplikace ergotaminu či dihydroergotaminu, není přesně známo, v případě nazální aplikace důkazy nasvědčují spíše bezpečnosti takových kombinací. Avšak držitelé rozhodnutí o registraci přípravků Migergot čípky, D.H.E. 45 nosní sprej a Migranal injekce v USA před souběžným podáváním silných inhibitorů CYP3A4 důrazně varují a považují je za kontraindikované [7,8].
Přehled léčiv, která patří mezi inhibitory CYP3A4, uvádíme v tabulce 2, v tabulce 3 pak přehled induktorů CYP3A4.
Souběžné podávání námelových
alkaloidů se silnými a středně silnými inhibitory CYP3A4 je
nezbytné považovat za rizikové, a je třeba se mu
vyhnout. V případě ergotaminu a dihydroergotaminu je
dokonce považováno za kontraindikované.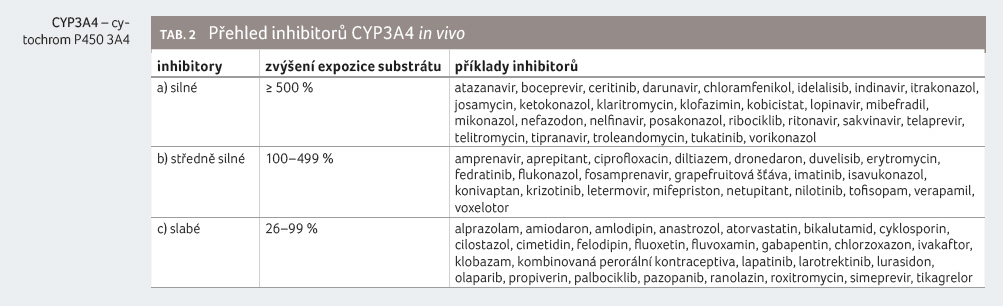
Souběžné podávání námelových
alkaloidů se silnými a středně silnými induktory CYP3A4 je
spojeno s rizikem selhání terapie, a je třeba se mu
vyhnout.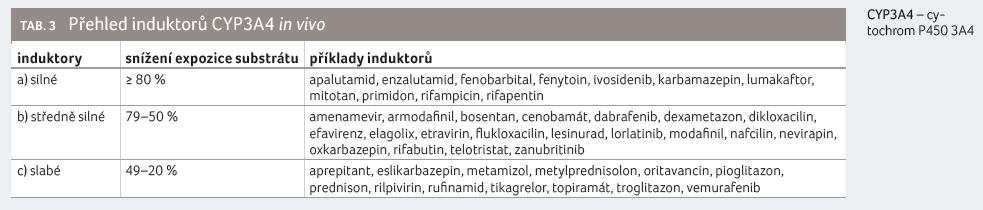
Námelové alkaloidy se kvůli navozené
vazokonstrikci nesmějí podávat současně s léky, které
mají vazokonstrikční účinek. Mezi takové léky patří zejména
triptany. To z důvodu předběžné opatrnosti vyžaduje
dodržovat bezpečnostní intervaly odstupů podání jednotlivých
léčiv. Například v případě sumatriptanu je doporučeno
před jeho užitím počkat nejméně 24 hodin po užití
ergotaminu nebo jiného triptanu. Naopak je doporučeno počkat
nejméně šest hodin po užití sumatriptanu před užitím
ergotaminu a nejméně 24 hodin před užitím jiného triptanu.
Konkrétní časové odstupy uvádí tabulka 4.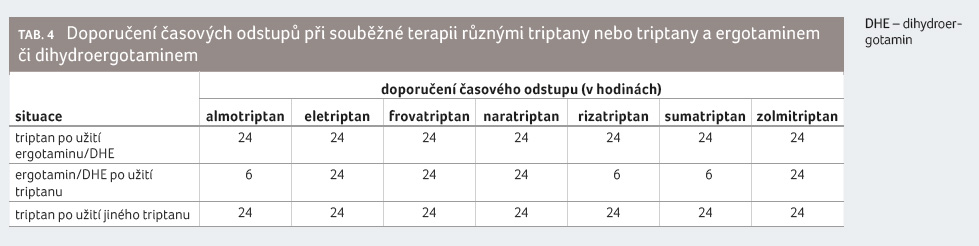
Lékové interakce triptanů
Triptany v současnosti již patří mezi klasická antimigrenika. Tvoří na první pohled homogenní skupinu léčiv s totožným nebo velmi blízkým mechanismem účinku (selektivní agonisté působící na serotoninovém 5 HT1B/1D receptoru, pouze eletriptan vykazuje též relevantní 5 HT1F agonistickou aktivitu). Mechanismus účinku triptanů souvisí jak s jejich klinickým účinkem, tak s jejich nežádoucími účinky, zejména s vazokonstrikcí (danou agonistickým účinkem na serotoninovém 5 HT1B/1D receptoru).
Farmakokinetické lékové interakce triptanů
Triptany mají odlišný osud v organismu daný odlišnými cestami své biotransformace, a proto mají odlišné farmakokinetické lékové interakce. Na metabolismu triptanů se spolupodílejí různé izoenzymy cytochromu P450 a monoaminooxidáza A (MAO A).
Sumatriptan je majoritně (> 80 %) metabolizován cestou MAO A, a to za vzniku farmakologicky neaktivního metabolitu [9]. To vysvětluje klinicky významné šestinásobné zvýšení expozice sumatriptanu v případě jeho souběžného podávání se selektivním inhibitorem MAO A moklobemidem [10,11].
Naratriptan se metabolizuje jen částečně, mírně nad 50 % podané dávky se vylučuje močí v nezměněné formě, z malé části je metabolizován CYP1A2 a CYP3A4 a nelze vyloučit, že patrně za účasti flavinové monooxygenázy (FMO1 nebo FMO3) [12]. Naratriptan tak s největší pravděpodobností nepředstavuje riziko lékových interakcí na podkladě ovlivnění jejich metabolismu.
Zolmitriptan se metabolizuje převážně (40 %) cestou CYP1A2, částečně (10−20 %) též cestou MAO A, přičemž hlavním metabolitem vznikajícím cestou CYP1A2 je N desmethyl zolmitriptan [13]. In vitro fluvoxamin již ve velmi nízkých koncentracích silně inhibuje tvorbu N desmethyl zolmitriptanu [14]. Souběžné podávání selektivního inhibitoru MAO A moklobemidu zvýšilo expozici zolmitriptanu pouze o 48 %, přičemž současně zvýšilo tvorbu N desmethyl zolmitriptanu o 105 % (tj. zvýšilo metabolizaci alternativní cestou CYP1A2).
Rizatriptan se metabolizuje majoritně (> 60 %) cestou MAO A, zdá se, že z malé části je též metabolizován cestou MAO B [15,16]. Souběžné podávání inhibitoru MAO A moklobemidu zvyšuje expozici rizatriptanu o 121 % a jeho hlavního metabolitu (N desmethyl rizatriptanu) o 425 % [17].
Almotriptan se metabolizuje jen částečně, přibližně 50 % podané dávky se vylučuje močí v nezměněné formě [18]. Biotransformace probíhá z větší části cestou MAO A a podílí se na ní také CYP3A4 a CYP2D6 [19]. Inhibitor MAO A moklobemid zvyšuje při souběžném podávání expozici almotriptanu pouze o 37 %, silný inhibitor CYP3A4 ketokonazol pak expozici almotriptanu zvyšuje o 57 % [20,21]. Studie prokazující lékovou interakci s inhibitory CYP2D6 dosud nebyla provedena.
Eletriptan je jediným triptanem, který se majoritně (> 80 %) metabolizuje cestou CYP3A4 a z malé části (kolem 10 %) pak cestou CYP2D6, proto nemůže překvapit, že silné inhibitory CYP3A4 při souběžném podávání zvyšují expozici eletriptanu na 491 % a středně silné inhibitory CYP3A4 zvyšují expozici eletriptanu na 103−174 % [22,23]. V experimentální studii u potkanů bylo prokázáno, že je li eletriptan aplikován podkožně, nevede jeho souběžné podávání s inhibitory CYP3A4 ke klinicky významné lékové interakci, z čehož plyne, že k lékové interakci dochází zejména cestou inhibice intestinální CYP3A4 [24].
Frovatriptan se částečně (< 20 %) metabolizuje cestou CYP1A2 [25]. Silný inhibitor CYP1A2 zvyšuje expozici souběžně podávanému frovatriptanu pouze o 39 % (u mužů) a o 41 % (u žen) [26].
Triptany tak lze z hlediska farmakokinetických lékových interakcí rozdělit na tři skupiny:
Triptany s minimálním nebo malým rizikem farmakokinetických lékových interakcí v důsledku inhibice nebo indukce jejich metabolismu, mezi něž patří:
- almotriptan (MAO A , CYP3A4 , CYP2D6 ),
- frovatriptan (MAO A 0, CYP1A2 až ),
- naratriptan (MAO A , CYP1A2 , CYP3A4 ),
- zolmitriptan (MAO A , CYP1A2 ).
V této skupině triptanů k lékovým interakcím v důsledku inhibice nebo indukce metabolismu sice dochází, ale tyto lékové interakce nejsou klinicky významné a mohou se uplatnit pouze výjimečně u konkrétních pacientů, např. s genetickým polymorfismem příslušného enzymu nebo alternativní metabolické cesty.
Triptany metabolizované z více než 60 % cestou CYP3A4:
- eletriptan ().
- Eletriptan se dle vyjádření držitele rozhodnutí o registraci v ČR nesmí podávat společně se silnými inhibitory CYP3A4.
- rizatriptan (),
- sumatriptan ().
Sumatriptan i rizatriptan jsou podle vyjádření držitelů rozhodnutí o registraci kontraindikovány při současném podávání silného inhibitoru MAO A moklobemidu.
Shrnutí výše uvedených informací uvádí tabulka 5.
V souvislosti s inhibicí
MAO A je nezbytné doplnit, že řada „přírodních“
látek má klinicky významnou MAO A inhibiční aktivitu.
Serotoninový syndrom jako důsledek inhibice MAO A tak byl
pozorován při kombinaci serotonergně působících léků, pokud
byly souběžně podávány extrakty z třezalky tečkované
(Hypericum perforatum), harmaly mnohodílné (Peganum
harmala) nebo rozchodnice růžové (Rhodiola rosea)
[27−29].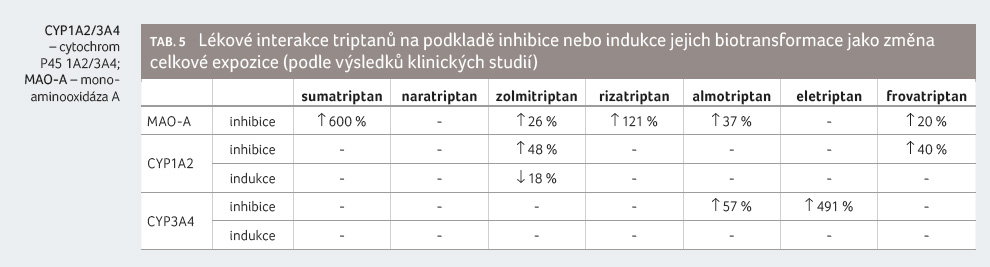
S velkou pravděpodobností jsou všechny triptany substráty transportního systému organických kationtů (organic cation transporter 1, OCT1), zejména při jejich vychytávání hepatocyty, i když důkazy jsou k dispozici pouze pro sumatriptan, naratriptan, rizatriptan a zolmitriptan, a to ještě ze studií in vitro [30]. Z klinického hlediska je zatím pouze známo, že genetický polymorfismus OCT1 (v případě jedinců se ztrátou transportní aktivity OCT1, např. polymorfismy rs12208357, rs7255763, rs34130495 nebo 34059508) je spojen s dvojnásobnou expozicí sumatriptanu. Zda souběžné podávání inhibitorů OCT1 bude mít obdobný efekt, není dosud známo, je to však pravděpodobné, zejména u nejsilnějších inhibitorů OCT1, jako jsou emtricitabin či verapamil [31,32].
Vzhledem k tomu, že OCT1 je
odpovědný za transport triptanů do jater (zde se pak
triptany biotransformují) a do CNS (zde triptany mají
farmakodynamický efekt), je role OCT1 patrně velmi důležitá
nejen z hlediska jejich farmakokinetických vlastností, ale
i z hlediska jejich klinické účinnosti. Inhibice OCT1 by
totiž mohla přispívat ke snížení účinku triptanů, jak
ostatně bylo prokázáno pro jiný substrát OCT1 lamotrigin [33].
Přehled inhibitorů OCT1 uvádí tabulka 6.
Klinické studie prokazující lékové interakce inhibitorů OCT1 s jinými léky, než jsou triptany, jsou již k dispozici. Například inhibice intestinální OCT1 vede ke zvýšení výskytu gastrointestinální intolerance metforminu [34]. V případě inhibitorů protonové pumpy autoři zjistili, že současné užívání metforminu a inhibitorů protonové pumpy vede ke zvýšení rizika gastrointestinální intolerance metforminu 1,84krát (1,32−2,57krát na 95% hladině spolehlivosti; p = 0,011).
Farmakodynamické lékové interakce
triptanů
Vazokonstrikční působení triptanů jsme již zmínili v souvislosti s jejich souběžným podáváním s námelovými alkaloidy.
Díky mechanismu účinku mají triptany serotonergní efekt a jejich kombinace s jinými serotonergními léky může vést ke zvýšení rizika vzniku serotoninového syndromu [35]. Triptany jsou naštěstí užívány intermitentně, a nadto obecně mají nižší riziko serotonergního působení, a proto uvedené riziko naštěstí není příliš vysoké [36].
FDA (FDA Information for Healthcare Professionals) přezkoumal 27 zpráv hlášení serotoninového syndromu při současném užívání antidepresiv ze skupiny selektivních inhibitorů zpětného vychytávání serotoninu (SSRI) nebo inhibitorů zpětného vychytávání serotoninu a noradrenalinu (SNRI) a triptanů. Ve dvou případech se jednalo o život ohrožující nežádoucí účinek, v dalších 13 případech byla nutná hospitalizace pacientů. Projevy serotoninového syndromu byly velmi variabilní a zahrnovaly respirační selhání, bezvědomí, mánie, halucinace, zmatenost, závratě, hypertermii, hypertenzi, pocení, třes, slabost a ataxii. V celkem osmi případech došlo ke vzniku serotoninového syndromu po nedávném zvýšení dávky nebo po zahájení terapie dalším serotonergním léčivem (SSRI, resp. SNRI nebo triptanem). Medián doby vzniku serotoninového syndromu po změně dávky nebo po zahájení terapie dalším serotonergním léčivem byl 24 hodin, s rozptylem 10 minut až šest dnů. Následně byly dostupné informace shrnuty a podrobeny přezkoumání a bylo konstatováno, že z celkem 40 kazuistik, které se týkaly výskytu serotoninového syndromu při současném podávání antidepresiv SSRI/SNRI a triptanů a byly publikovány v odborné literatuře nebo shromážděny FDA, pouze 10 splňuje Sternbachova diagnostická kritéria pro serotoninový syndrom a žádná z nich pak Hunterova kritéria pro serotoninovou toxicitu, resp. nebyla k dispozici náležitá dokumentace [37]. Ačkoliv podle autorů neexistují jednoznačné důkazy pro potvrzení lékové interakce, je vzhledem k závažnosti serotoninového syndromu namístě zvýšená opatrnost.
K naprosté většině případů serotoninového syndromu (84 %) dochází při zahájení terapie druhým serotonergním léčivem nebo při zvýšení jeho dávkování. Z tohoto důvodu se někteří autoři domnívají, že současnému podávání tramadolu a antidepresiv je nutné se zcela vyhnout [38]. Před současným podáváním antimigrenik ze skupiny triptanů a opioidních analgetik a tramadolu varují Ansari a kol., podle nichž je potenciál vzniku serotoninového syndromu při takové kombinaci značný [39]. Podobně lze předpokládat zvýšené riziko serotoninového syndromu v případě, že jsou triptany souběžně podávány s inhibitory MAO A nebo s neselektivními inhibitory MAO (např. s linezolidem). Důvodem je souběh farmakokinetické lékové interakce (zvýšení expozice všem triptanům s výjimkou naratriptanu a eletriptanu) a farmakodynamické lékové interakce (zvýšení serotonergní aktivity).
Seznam použité literatury
- [1] Námelové alkaloidy – omezení použití. SÚKL 8. 7. 2013. Dostupné na: https://www.sukl.cz/namelove‑alkaloidy‑omezeni‑pouziti?highlightWords=namelove+alkaloidy
- [2] Couet W, Mathieu HP, Fourtillan JB. Effect of Ponsinomycin on the Pharmacokinetics of Dihydroergotamine Administered Orally. Fundam Clin Pharmacol 1991; 5: 47–52.
- [3] Althaus M, Retzow A, Castell JV, et al. In vitro identification of the cytochrome P450 isoform responsible for the metabolism of alpha‑dihydroergocryptine. Xenobiotica 2000; 30: 1033–1045.
- [4] FDA Adverse Event Reporting System (FAERS) Public Dashboard, FDA, USA, 2021. Dostupné na: https://www.fda.gov/drugs/fda‑adverse‑event‑reporting‑system‑faers/fda‑adverse‑event‑reporting‑system‑faers‑public‑dashboard
- [5] de Mey C, Althaus M, Ezan E, Retzow A. Erythromycin increases plasma concentrations of alpha‑dihydroergocryptine in humans. Clin Pharmacol Ther 2001; 70: 142–148.
- [6] Kellerman D, Kori S, Forst A, et al. Lack of drug interaction between the migraine drug MAP0004 (orally inhaled dihydroergotamine) and a CYP3A4 inhibitor in humans. Cephalalgia 2012; 32: 150–158.
- [7] FDA Full Prescribing Information: D.H.E. 45® (dihydroergotamin), 9/2009. Dostupné na: https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/005929s044lbl.pdf
- [8] FDA Full Prescribing Information: Migranal® (dihydroergotamin), 8/2019. Dostupné na: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/020148Orig1s025lbl.pdf
- [9] Dixon CM, Park GR, Tarbit MH. Characterization of the enzyme responsible for the metabolism of sumatriptan in human liver. Biochem Pharmacol 1994; 47: 1253–1257.
- [10] Blier P, Bergeron R. The safety of concomitant use of sumatriptan and antidepressant treatments. J Clin Psychopharmacol 1995; 15: 106–109.
- [11] FDA Full Prescribing Information: Imitrex® (sumatriptan), 7/2018. Dostupné na: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/020080s052lbl.pdf
- [12] FDA Full Prescribing Information: Amerge® (naratriptan), 11/2016. Dostupné na: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/020763s011lbl.pdf
- [13] Wild MJ, McKillop D, Butters CJ. Determination of the human cytochrome P450 isoforms involved in the metabolism of zolmitriptan. Xenobiotica 1999; 29: 847–857.
- [14] Yu LS, Yao TW, Zeng S. In vitro metabolism of zolmitriptan in rat cytochromes induced with beta‑naphthoflavone and the interaction between six drugs and zolmitriptan. Chem Biol Interact 2003; 146: 263–272.
- [15] Iwasa T, Sano H, Sugiura A, et al. An in vitro interethnic comparison of monoamine oxidase activities between Japanese and Caucasian livers using rizatriptan, a serotonin receptor 1B/1D agonist, as a model drug. Br J Clin Pharmacol 2003; 56: 537–544.
- [16] Ucar G. Interaction of Rizatriptan with Rat Liver Monoamine Oxidases. Hacettepe University, Journal of Faculty of Pharmacy 2004; 24: 25–35.
- [17] van Haarst AD, Gerven JMA, Cohen AF, et al. The effects of moclobemide on the pharmacokinetics of the 5‑HT1B/1D agonist rizatriptan in healthy volunteers. Br J Clin Pharmacol 1999; 48: 190–196.
- [18] Buzzi MG. Pathways to the best fit of triptans for migraine patients. Cephalalgia 2008; 28 (Suppl 2): 21–27.
- [19] McEnroe JD, Fleishaker JC. Clinical pharmacokinetics of almotriptan, a serotonin 5‑HT(1B/1D) receptor agonist for the treatment of migraine. Clin Pharmacokinet 2005; 44: 237–246.
- [20] Fleishaker JC, Ryan KK, Jansat JM, et al. Effect of MAO‑A inhibition on the pharmacokinetics of almotriptan, an antimigraine agent in humans. Br J Clin Pharmacol 2001; 51: 437–441.
- [21] Fleishaker JC, Herman BD, Carel BJ, Azie NE. Interaction between ketoconazole and almotriptan in healthy volunteers. J Clin Pharmacol 2003; 43: 423–427.
- [22] Kim YK, Shin KH, Alderman J, et al. Pharmacokinetics and tolerability of eletriptan hydrobromide in healthy Korean subjects. Drug Des Devel Ther 2018; 12: 331–337.
- [23] FDA Full Prescribing Information: Relpax® (eletriptan), 3/2020. Dostupné na: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/21016s029lbl.pdf
- [24] Patel H, Desai N, Patel P, et al. Differential pharmacokinetic drug‑drug interaction potential of eletriptan between oral and subcutaneous routes. Xenobiotica 2019; 49: 1202–1208.
- [25] Balbisi EA. Frovatriptan: a review of pharmacology, pharmacokinetics and clinical potential in the treatment of menstrual migraine. Ther Clin Risk Manag 2006; 2: 303–308.
- [26] Buchan P, Wade A, Ward C, et al. Frovatriptan has no clinically significant interaction with fluvoxamine. Cephalalgia 2001; 21: 427.
- [27] Bonetto N, Santelli L, Battistin L, Cagnin A. Serotonin syndrome and rhabdomyolysis induced by concomitant use of triptans, fluoxetine and hypericum. Cephalalgia 2007; 27: 1421–1423.
- [28] Bakim B, Sertcelik S, Tankaya O. A Case of Serotonin Syndrome with Antidepressant Treatment and Concomitant use of The Herbal Remedy (Peganum Harmala). Bull Clin Psychopharmacol 2012; 22: 359–361.
- [29] Maniscalco I, Toffol E, Giupponi G, Conca A. The interaction of Rhodiola rosea and antidepressants. A case report. Neuropsychiatr 2015; 29: 36–38.
- [30] Matthaei J, Kuron D, Faltraco F, et al. OCT1 mediates hepatic uptake of sumatriptan and loss‑of‑function OCT1 polymorphisms affect sumatriptan pharmacokinetics. Clin Pharmacol Ther 2016; 99: 633–641.
- [31] Zeng Q, Bai M, Li C, et al. Multiple Drug Transporters Contribute to the Placental Transfer of Emtricitabine. Antimicrob Agents Chemother 2019; 63: e00199–e00219.
- [32] Ahlin G, Karlsson J, Pedersen JM, et al. Structural requirements for drug inhibition of the liver specific human organic cation transport protein 1. J Med Chem 2008; 51: 5932–5942.
- [33] Dickens D, Owen A, Alfirevic A, et al. Lamotrigine Is a Substrate for OCT1 in Brain Endothelial Cells. Biochem Pharmacol 2012; 83: 805–814.
- [34] Dujic T, Zhou K, Donnelly LA, et al. Association of Organic Cation Transporter 1 With Intolerance to Metformin in Type 2 Diabetes: A GoDARTS Study. Diabetes 2015; 64: 1786–1793.
- [35] Fine A, Bastings E. Triptans and serotonin syndrome. Headache 2012; 52: 1184–1185.
- [36] Culbertson VL, Rahman SE, Bosen GC, et al. Implications of Off‑Target Serotoninergic Drug Activity: An Analysis of Serotonin Syndrome Reports Using a Systematic Bioinformatics Approach. Pharmacotherapy 2018; 38: 888–898.
- [37] Ewans RW, Tepper SJ, Shapiro RE, et al. The FDA alert on serotonin syndrome with use of triptans combined with selective serotonin reuptake inhibitors or selective serotonin‑norepinephrine reuptake inhibitors: American Headache Society position paper. Headache 2010; 50: 1089–1099.
- [38] Park SH, Wackernah RC, Stimmel GL, et al. Serotonin syndrome: is it a reason to avoid the use of tramadol with antidepressants? J Pharm Pract 2014; 27: 71–78.
- [39] Ansari H, Kouti L. Drug Interaction and Serotonin Toxicity with Opioid Use: Another Reason to Avoid Opioids in Headache and Migraine Treatment. Curr Pain Headache Rep 2016; 20: 50.