Acalabrutinib
Souhrn:
Lapka M. Acalabrutinib. Remedia 2021; 31: 54–62.
Acalabrutinib je vysoce selektivní, ireverzibilní a perorálně podávaný inhibitor proteinkináz druhé generace. Látka je selektivním inhibitorem Brutonovy kinázy a je aktuálně schválena v monoterapii nebo v kombinaci s obinutuzumabem v indikaci k léčbě dospělých s dříve neléčenou chronickou lymfocytární leukemií (CLL) a k léčbě dospělých jedinců s CLL, kteří podstoupili alespoň jednu předchozí terapii. Současně probíhá velké množství klinických studií, které by měly ujasnit možnou roli acalabrutinibu v dalších indikacích, případně doplnit další informace v indikacích již schválených.
Summary:
Lapka M. Acalabrutinib. Remedia 2021; 31: 54–62.
Acalabrutinib is a highly selective, irreversible and orally administered 2nd generation protein kinase inhibitor. The substance is a selective inhibitor of Brutonʼs tyrosine kinase and is currently approved as a monotherapy or in a combination with obinutuzumab for the treatment of adults with previously untreated chronic lymphocytic leukaemia (CLL) and for the treatment of adults with CLL who have received at least one prior therapy. At the same time, a large number of clinical studies are ongoing to clarify the possible role of acalabrutinib in other indications or to amend additional information in already approved indications.
Key words: acalabrutinib, Brutonʼs kinase inhibitor, cytostatic, chronic lymphocytic leukaemia
Úvod
Chronická lymfocytární leukemie (CLL) je nejčastěji se vyskytující leukemií v západním světě a představuje nejčastější leukemii u dospělých pacientů. Onemocnění se rozvíjí převážně u starších pacientů s mediánem věku 70 let při stanovení diagnózy. Etiologie CLL buněk je odvozována z vývojových stadií B lymfocytů a předpokládá se, že kromě antigenní stimulace hraje v patogenezi CLL roli narušení apoptózy nebo prostředí [1].
Léčba CLL se neustále nachází v procesu vývoje. Dříve byla terapie zaměřena na potlačení symptomů, aktuálně však lze s využitím kombinované imunoterapie přivodit kompletní remisi onemocnění. Intenzivní léčba je vhodná pro fyzicky zdatné pacienty, CLL se však převážně vyskytuje u starší populace, a tím spíše je vždy nutno zvážit prospěch z terapie [2].
V poslední době je s léčbou CLL sledován proces buněčné signalizace přes B buněčný receptor (BCR), jehož součástí je Brutonova tyrozinkináza (BTK). Klinické studie prokázaly, že perorálně podávané selektivní a ireverzibilní inhibitory této kinázy inhibují BCR signalizaci a dokáží indukovat cílenou apoptózu. Přípravek Calquence (acalabrutinib) byl registrován Evropskou lékovou agenturou (EMA) dne 5. listopadu 2020 a v České republice aktuálně (leden 2021) ještě není dostupný z důvodu nestanovené úhrady.
Acalabrutinib je řazen do farmakoterapeutické skupiny Cytostatika, inhibitor proteinkináz, ATC kód L01XE51. Léčivý přípravek Calquence slouží k perorálnímu podání ve formě tvrdých tobolek obsahujících acalabrutinib v síle 100 mg. Doporučená dávka je 100 mg acalabrutinibu dvakrát denně (odpovídá celkové denní dávce 200 mg).
Léčivo je bílý až žlutý prášek s rozpustností závislou na hodnotě pH. Látka je rozpustná ve vodě při hodnotě pH < 3 a je prakticky nerozpustná ve vodě při pH > 6, je rozpustná v organických rozpouštědlech, jako je etanol, dimetylsulfoxid a dimetylformamid. Acalabrutinib je chemicky podle IUPAC (International Union of Pure and Applied Chemistry) 4 [8 amino 3 [(2S) 1 but 2 ynoylpyrrolidin 2 yl]imidazo[1,5 a]pyrazin 1 yl] N pyridin 2 ylbenzamid, sumární vzorec: C26H23N7O2. Jeho molekulová hmotnost činí 465,5 g/mol.
Farmakodynamika
Acalabrutinib představuje vysoce selektivní, ireverzibilní a perorálně podávaný inhibitor BTK druhé generace prostřednictvím kovalentní vazby Cys481 u pacientů s CLL. Brutonova tyrozinkináza je signální molekula BCR a drah cytokinových receptorů. Signalizace BTK v B buňkách vede k jejich přežití a proliferaci a je nutná pro buněčnou adhezi, prostup a chemotaxi. Acalabrutinib a jeho aktivní metabolit, ACP 5862, vytvářejí kovalentní vazbu s cysteinovým reziduem (Cys481) v aktivním místě BTK, což vede k ireverzibilní inaktivaci BTK s minimálními interakcemi mimo cílovou oblast [3].
Účinky acalabrutinibu na CLL buňky, T buňky, NK buňky a epiteliální buňky byly studovány signálními a funkčními testy. Acalabrutinib inhiboval purifikovanou BTK s IC50 (koncentrace inhibitoru, která způsobí 50% inhibici enzymu) 3 nM a EC50 (střední účinná koncentrace) 8 nM v testu CD69 aktivace B lymfocyty [3].
Preklinický výzkum
V signálním testu in vitro na primárních humánních CLL buňkách acalabrutinib inhiboval tyrozinovou fosforylaci kináz ERK, IKB a AKT. Acalabrutinib prokázal vyšší BTK selektivitu stanovením IC50 na devíti kinázách s cysteinovým zbytkem ve stejné poloze jako BTK. Důležité je, že na rozdíl od jiných acalabrutinib neinhibuje receptor epidermálního růstového faktoru (EGFR), interleukinem indukovatelnou T buněčnou kinázu (ITK) nebo tyrozinovou proteinkinázu (TEC). V testech in vitro bylo jasně prokázáno, že na rozdíl od ibrutinibu neměl acalabrutinib žádný účinek na fosforylaci EGFR na tyrozinových zbytcích y1068 a y1173. Ve srovnání s ibrutinibem má acalabrutinib mnohem vyšší IC50 (> 1 000 nM) nebo prakticky žádnou inhibici kinázových aktivit ITK, EGFR, ERBB2, ERBB4, JAK3, BLK, FGR, FYN, HCK, LCK, LYN, SRC a YES1 [4].
Acalabrutinib byl in vivo hodnocen na modelech B buněčného nehodgkinského lymfomu (NHL). Ve studii modelu B buněk NHL byl perorálně podáván acalabrutinib v eskalujících dávkách 2,5 mg/kg každých 24 hodin (6 zvířat), 5 mg/kg každých 24 hodin (5 zvířat) nebo 10 mg/kg každých 12 hodin (1 zvíře). Ve výsledku tři zvířata dosáhla částečné remise (partial remission, PR), tři dosáhla stabilního onemocnění (stable disease, SD), zatímco zbývajících šest zvířat mělo progresi onemocnění (progressive disease, PD) [5].
Účinky acalabrutinibu proti buňkám CLL byly demonstrovány na myším modelu s xenografty lidské CLL. Acalabrutinib významně a závisle na dávce inhiboval proliferaci lidských CLL buněk ve slezinách myší ve všech dávkách, měřeno pro expresi Ki67 (p = 0,002) [6], a v dalším in vivo modelu léčba acalabrutinibem významně zvýšila přežití proti placebu (medián 81 vs. 59 dnů, p = 0,02) [7].
U pacientů, kteří trpěli malignitami B buněk a užívali acalabrutinib v dávce 100 mg dvakrát denně, byl medián obsazenosti BTK v ustáleném stavu ≥ 95 % v periferní krvi po dobu více než 12 hodin, což vedlo k inaktivaci BTK v průběhu doporučeného dávkovacího intervalu.
Farmakokinetika
Farmakokinetika acalabrutinibu a jeho aktivního metabolitu (ACP 5862) byly studovány u zdravých dobrovolníků a pacientů s malignitami B buněk. Acalabrutinib vykazuje dávkovou proporcionalitu a obě aktivní látky vytvářejí téměř lineární kinetiku v rozmezí dávek od 75 do 250 mg.
Absorpce
Při doporučeném dávkování 100 mg dvakrát denně u pacientů s malignitami B buněk geometrický průměr plochy pod křivkou plazmatické koncentrace v závislosti na čase v ustáleném stavu v průběhu 24 hodin (AUC24h) a maximální plazmatická koncentrace (cmax) pro acalabrutinib činily 1 843 ng·hr/ml, resp. 563 ng/ml, a pro ACP 5862 činily 3 947 ng·hr/ml, resp. 451 ng/ml.
Čas dosažení maximální plazmatické koncentrace (tmax) byl 0,5–1,5 hodiny (průměrná hodnota 0,9 h, medián 0,75 h) u acalabrutinibu a 1,0 hodiny pro ACP 5862 (jiná data hodnotí 1,6 h). Průměrná absolutní biodostupnost byla 25 %. Medián vázání na BTK v ustáleném stavu je z více než 95 % udržován v intervalu 12 hodin.
Vliv jídla
U zdravých subjektů podání jednotlivé dávky 75 mg s vysokokalorickým jídlem s vysokým obsahem tuku neovlivnilo průměrnou AUC v porovnání s podáním nalačno. Výsledná cmax však byla snížena o 69‒73 % a tmax byl opožděn o 1–2 hodiny.
Distribuce
Reverzibilní vazba na plazmatické proteiny byla 97,5 % u acalabrutinibu a 98,6 % u ACP 5862. Průměrný poměr krve a plazmy in vitro činil 0,8 u acalabrutinibu a 0,7 u ACP 5862. Průměrný distribuční objem v ustáleném stavu (Vss) byl u acalabrutinibu přibližně 34 litrů.
Biotransformace/metabolismus
In vitro je acalabrutinib metabolizován převážně prostřednictvím enzymů cytochromu P450 3A (CYP3A) a v menším rozsahu konjugací s glutationem a hydrolýzou amidů. Aktivní metabolit byl identifikován jako hlavní metabolit v plazmě, který je metabolizován primárně oxidací skrze CYP3A s AUC přibližně dvoj až trojnásobně vyšší ve srovnání s acalabrutinibem. ACP 5862 je přibližně o 50 % méně účinný při inhibici BTK ve srovnání s acalabrutinibem.
In vitro studie naznačují, že acalabrutinib v klinicky relevantních koncentracích neinhibuje CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, UGT1A1 nebo UGT2B7 a pravděpodobně neovlivňuje clearance substrátů pro tyto CYP. In vitro studie naznačují, že ACP 5862 v klinicky relevantních koncentracích neinhibuje CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4/5, UGT1A1 nebo UGT2B7 a pravděpodobně neovlivňuje clearance substrátů pro tyto CYP.
Interakce s transportními proteiny
In vitro studie naznačují, že acalabrutinib a ACP 5862 jsou substráty pro P glykoprotein (P gp) a protein rezistence karcinomu prsu (BCRP). Souběžné podávání s inhibitorem přenašeče organických aniontů OATP1B1/1B3 (600 mg rifampicinu, jednorázová dávka) vedlo ke zvýšení cmax, resp. AUC acalabrutinibu 1,2násobně, resp. 1,4násobně (n = 24, zdraví dobrovolníci), což není klinicky relevantní.
Acalabrutinib a ACP 5862 v klinicky relevantních koncentracích neinhibují P gp, OAT1, OAT3, OCT2, OATP1B1, OATP1B3 a MATE2 K. V klinicky relevantních koncentracích může acalabrutinib inhibovat střevní BCRP, zatímco ACP 5862 může inhibovat MATE1 (mnohočetný lékový a toxinový extruzní transportér 1). V klinicky relevantních koncentracích acalabrutinib neinhibuje MATE1, zatímco ACP 5862 neinhibuje BCRP.
Eliminace
Po jedné perorálně podané dávce 100 mg acalabrutinibu byl terminální poločas eliminace (t1/2) 1‒2 hodiny a t1/2 jeho aktivního metabolitu činil přibližně sedm hodin. U pacientů s malignitami B buněk byla průměrná zdánlivá perorální clearance (CL/F) 134 l/h v případě acalabrutinibu a 22 l/h v případě ACP 5862. Po podání jedné dávky 100 mg radioaktivně značeného [14C] acalabrutinibu bylo u zdravých subjektů 84 % dávky vyloučeno stolicí a 12 % dávky bylo vyloučeno močí. Méně než 2 % dávky bylo vyloučeno jako nezměněný acalabrutinib.
Na základě farmakokinetické analýzy populace bylo zjištěno, že věk (> 18 let), pohlaví, rasa a tělesná hmotnost nemají klinicky významný vliv na farmakokinetiku acalabrutinibu a jeho aktivního metabolitu. U pacientů ve věku do 18 let nebyly provedeny žádné farmakokinetické studie.
Acalabrutinib je minimálně vylučován ledvinami. Farmakokinetická studie s pacienty s poruchou funkce ledvin nebyla provedena. Na základě populační farmakokinetické analýzy nebyl u 408 pacientů s lehkou poruchou funkce ledvin (odhadovaná glomerulární filtrace [eGFR] 60–89 ml/min/1,73 m2, hodnoceno podle rovnice MDRD [Modification of Diet in Renal Disease]), u 109 pacientů se středně těžkou poruchou funkce ledvin (eGFR 30–59 ml/min/1,73 m2) v porovnání se 192 pacienty s normální funkcí ledvin (eGFR ≥ 90 ml/min/1,73 m2) pozorován žádný klinicky relevantní rozdíl. Farmakokinetika acalabrutinibu nebyla popsána u pacientů s těžkou poruchou funkce ledvin (eGFR < 29 ml/min/1,73 m2) nebo s poruchou ledvin vyžadující dialýzu. Pacienti s koncentracemi kreatininu vyššími než 2,5násobek institucionální horní hranice normy (ULN) nebyli do klinických studií zahrnuti.
Ve studiích s poruchami funkce jater v porovnání se subjekty s normální funkcí jater (n = 6) byla AUC acalabrutinibu zvýšena 1,9krát, resp. 1,5krát, resp. 5,3krát u subjektů s lehkou (n = 6; Childovo‒Pughovo skóre A), středně těžkou (n = 6; Childovo‒Pughovo skóre B), resp. těžkou (n = 8; Childovo‒Pughovo skóre C) poruchou funkce jater. Na základě analýzy nebyl pozorován rozdíl mezi subjekty s lehkou (n = 79) nebo středně těžkou (n = 6) poruchou funkce jater (celkový bilirubin mezi 1,5 a trojnásobkem ULN a kterékoliv aspartátaminotransferázy [AST]) oproti subjektům s normální (n = 651) funkcí jater (celkový bilirubin a AST v mezích ULN).
Klinické zkušenosti
K posouzení bezpečnosti, účinnosti, farmakokinetiky a farmakodynamiky acalabrutinibu u CLL byla iniciována multicentrická otevřená klinická studie fáze I/II s eskalovanými dávkami. Ve zprávě z roku 2016 bylo zařazeno 61 pacientů s relabující CLL. Ve fázi I byli pacienti léčeni acalabrutinibem ve zvyšující se dávce 100‒400 mg jednou denně. V expanzní fázi II bylo podáno 100 mg dvakrát denně. Po mediánu sledování 14,3 měsíce (rozmezí 0,5–20) byla celková míra odpovědi (objective response rate, ORR) 95 %, s 85% PR, 10% PR s lymfocytózou. U zbývajících 5 % pacientů byla hlášena SD.
Látka neinhibovala TEC a agregaci krevních destiček, což se nabízí jako výhodná volba oproti ibrutinibu při sníženém riziku krvácení. Acalabrutinib neinhiboval EGFR, takže byl pozorován nižší výskyt nežádoucích účinků (NÚ) na kůži a s ohledem na průjmy. Přechodné bolesti hlavy byly hlášeny jako častá příhoda [4].
Bezpečnost a účinnost
acalabrutinibu byly u dříve neléčené CLL hodnoceny
u dospělých pacientů v randomizované multicentrické
otevřené studii fáze III (ELEVATE TN) zahrnující 535
pacientů. Nemocní byli randomizováni do tří ramen:
acalabrutinib v monoterapii, acalabrutinib v kombinaci
s obinutuzumabem a obinutuzumab v kombinaci
s chlorambucilem. Primárním cílovým ukazatelem studie bylo
přežití bez progrese (progression free survival, PFS) podle
hodnocení IRC (Independent Review Comittee). IRC hodnocení PFS při
mediánu délky sledování pacientů 28,3 měsíce prokázalo 90%
snížení rizika progrese onemocnění nebo úmrtí při léčbě
kombinací acalabrutinib + obinutuzumab versus
obinutuzumb + chlorambucil. Medián PFS: při léčbě
kombinací acalabrutinib + obinutuzumab nedosažen (NR)
versus 22,6 měsíce (95% interval spolehlivosti [CI] 20,2‒27,6)
při léčbě kombinací obinutuzumab + chlorambucil.
Shodné zlepšení PFS bylo pozorováno při léčbě kombinací
acalabrutinib a acalabrutinib + obinutuzumab napříč
všemi podskupinami pacientů bez ohledu na vysoce rizikové
genomické charakteristiky (vysoké riziko definováno jako
nemutovaný IGHV, del17p, del11q, mutovaný TP53 nebo komplexní
karyotyp) [8].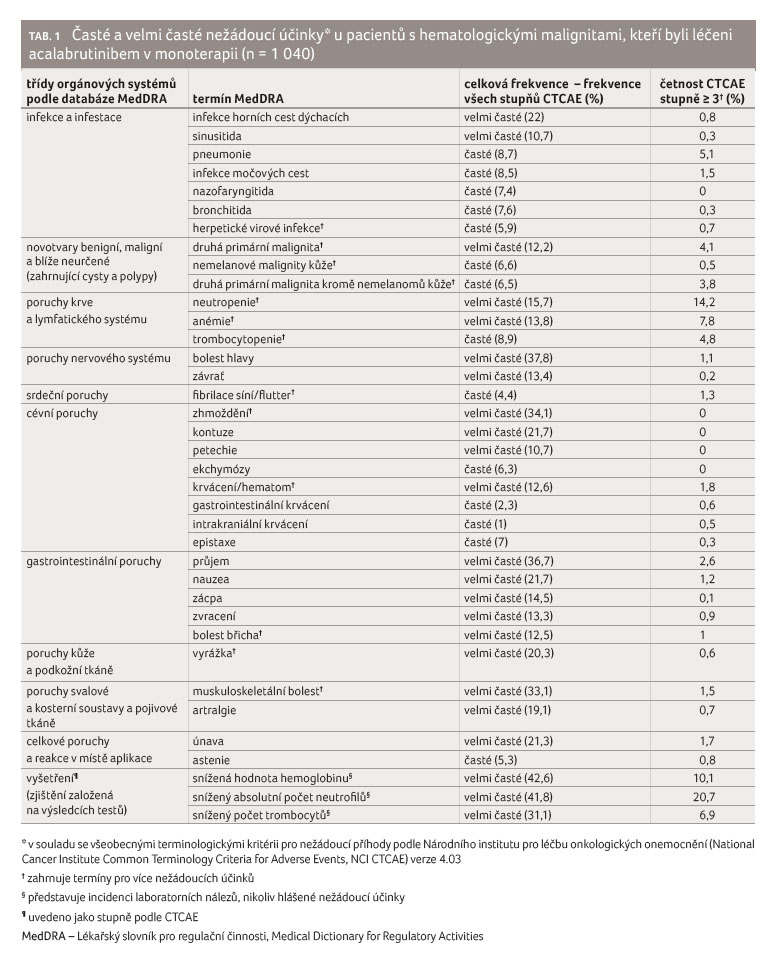
Randomizovaná multicentrická otevřená studie fáze III (ASCEND) zahrnující 310 dospělých pacientů, kteří v minulosti podstoupili alespoň jeden typ léčby kromě terapie inhibitory BCL 2 nebo inhibitory receptorů B buněk, hodnotila bezpečnost a účinnost acalabrutinibu u recidivující nebo refrakterní CLL. Primárním cílovým ukazatelem studie bylo PFS podle hodnocení IRC. Při mediánu sledování 16,1 měsíce bylo na podkladě PFS prokázáno 69% statisticky významné snížení rizika úmrtí nebo progrese onemocnění ve skupině s acalabrutinibem. Medián PFS: při léčbě acalabrutinibem nedosažen (NR) versus 16,5 měsíce (95% CI 14,0‒17,1) při léčbě režimem idelalisib + rituximab nebo bendamustin + rituximab [9].
Seznam častých a velmi častých NÚ ze skupiny zahrnující 1 040 pacientů léčených acalabrutinibem v monoterapii shrnuje tabulka 1.
Zpráva z roku 2020 popisuje dramatické zlepšení výsledků přežití u pacientů s CLL/malým lymfocytárním lymfomem (SLL) po podání cílené terapie a potvrzuje účinnost, trvání odpovědi a dlouhodobou bezpečnost. Ve studii fáze Ib/II dostávalo 134 pacientů s relabující/refrakterní (R/R) CLL nebo SLL acalabrutinib 100 mg dvakrát denně (věkový medián 66 let [rozmezí 42‒85 let]; medián počtu předchozích terapií 2 [rozmezí 1‒13]) po dobu mediánu 41 měsíců (rozmezí 0,2‒58 měsíců). Většina NÚ byla mírná nebo středně závažná a nejčastěji šlo o průjem (52 %) a bolest hlavy (51 %). Závažnějšími NÚ byly neutropenie (14 %), pneumonie (11 %), hypertenze (7 %), anémie (7 %) a průjem (5 %). Fibrilace síní a závažné krvácení (všechny stupně NÚ) se vyskytly u 7 %, resp. 5 % pacientů. Většina pacientů (56 %) setrvala v léčbě; primárními důvody přerušení byly PD (21 %) a NÚ (11 %) [11].
Celková míra odpovědi včetně částečné odpovědi s lymfocytózou byla u acalabrutinibu 94 %; odpovědi byly podobné bez ohledu na genomické rysy ‒ přítomnost del(11) (q22.3), del(17) (p13.1), komplexního karyotypu nebo stavu mutace těžkého řetězce variabilní oblasti imunoglobulinu. Nebylo dosaženo střední doby trvání odpovědi a PFS; odhadovaný 45měsíční PFS činil 62 % (95% CI 51‒71). Mutace BTK byla detekována u šesti z devíti pacientů (67 %) při relapsu [11].
Zajímavé výsledky prokázaly dosavadní studie zabývající se použitím inhibitorů BTK u pacientů s lymfomem z plášťových buněk (MCL). Celkem 80 % pacientů dosáhlo výborných výsledků a další běžící studie prozatím ukazují data podporující dosavadní schválenou indikaci při dosažení dobré tolerance a snášenlivosti léčiva [12].
Zařazení do současné palety
léčiv
Aktuální zahraniční doporučení NCCN (National Comprehensive Cancer Network) guidelines pro CLL verze 1.2021 uvádějí pro léčbu první linie CLL u dospělých pacientů jako doporučení důkazní kategorie 1 bez ohledu na věk, komorbidity a přítomnost delece 17p nebo mutace TP53 preferenčně tyto léčebné režimy: acalabrutinib ± obinutuzumab, ibrutinib v monoterapii a venetoklax + obinutuzumab. Pro léčbu R/R CLL u dospělých pacientů jako doporučení důkazní kategorie 1 bez ohledu na věk, komorbidity a přítomnost delece 17p nebo mutace TP53 preferenčně tyto léčebné režimy: acalabrutinib, ibrutinib a venetoklax + rituximab [18]. Acalabrutinib je již obsažen v doporučení ESMO (European Society for Medical Oncology) pro léčbu CLL i přes jeho teprve nedávnou registraci EMA. Acalabrutinib má dle ESMO, podobně jako ibrutinib a kombinace venetoklax + rituximab, nejvyšší míru doporučení IA [19].
Porovnání s jinými léčivy
ze skupiny
Na rozdíl od ibrutinibu se nezdá, že by byl acalabrutinib ireverzibilně zaměřen na jiné kinázy než BTK, včetně EGFR, TEC a ITK. Selektivní cílení na BTK pomocí acalabrutinibu může částečně vysvětlit rozdíly snášenlivosti pozorované mezi acalabrutinibem a ibrutinibem v dosavadních klinických studiích.
Ve srovnání s ibrutinibem prokázal acalabrutinib vyšší selektivitu a inhibici aktivity cílového BTK. Preklinické studie popsaly, že se jedná o účinnější a selektivnější inhibitor BTK ze své třídy. Zároveň vykázal acalabrutinib mnohem vyšší IC50 bez ovlivnění kinázových aktivit ITK, TEC, EGFR, ERBB2, ERBB4, JAK3, BLK, FGR, FYN, HCK, LCK, LYN, SRC a YES1. Navíc léčba ibrutinibem v dávce 420 mg jednou denně vedla k ovlivnění formování trombů, zatímco ve srovnání s kontrolou a pacienty léčenými acalabrutinibem v dávce 100 mg dvakrát denně nebyl zjištěn žádný vliv. Data z roku 2016 naznačují lepší bezpečnostní profil acalabrutinibu s minimálními nepříznivými účinky ve srovnání s ibrutinibem [13].
In vitro testy uvádějí, že acalabrutinib neměl žádný účinek na fosforylaci EGFR na tyrozinových zbytcích y1068 a y1173. V koncentraci 1 000 nM ibrutinib úplně inhiboval aktivitu TEC, nicméně 1 000 nM acalabrutinibu mělo na aktivitu TEC minimální vliv. Účinky ibrutinibu a acalabrutinibu na krevní destičky byly také srovnávány v modelu myší trombózy VWFHA1. U poškozených arteriol jedinců léčených ibrutinibem v dávce 420 mg jednou denně nebo acalabrutinibem v dávce 100 mg dvakrát denně byla hodnocena tvorba trombů. Velikost trombu ve skupině s acalabrutinibem byla srovnatelná se zdravou kontrolou, zatímco tvorba trombů byla u ibrutinibu jasně inhibována. Tato data naznačují, že acalabrutinib je na rozdíl od ibrutinibu selektivnější pro inhibici BTK a nemá prakticky žádný vliv na inhibici aktivity trombocytů [4].
Dávkování
Perorální podávání acalabrutinibu ve formě tobolek je doporučeno v dávkování dvakrát denně ideálně ve 12hodinovém rozmezí bez ohledu na příjem jídla. Tobolky by měly být spolknuty s dostatečným množstvím vody, neměly by být rozkousnuty, rozlomeny nebo žvýkány. Pokud pacient promešká podání dávky o více než tři hodiny, měl by tuto dávku vynechat a užít další podle naplánovaného rozvrhu. Pacient by neměl užívat tobolky navíc ani při vynechání dávky.
Perorálně by se mělo podávat 100 mg
dvakrát denně a pokračovat do PD nebo do rozvoje
nepřijatelné toxicity. Doporučené modifikace dávky podle NÚ
jsou uvedeny v tabulce 2.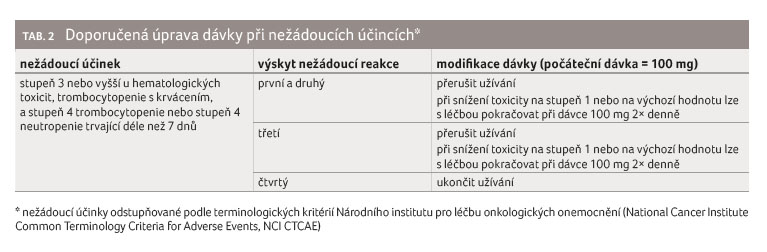
Souběžnému podávání acalabrutinibu se silnými inhibitory CYP3A4 je doporučeno se vyhnout. Pokud je zapotřebí zahájit krátkodobou léčbu kratší než sedm dnů, podávání acalabrutinibu by mělo být během terapie přerušeno. Pokud je souběžně podáván střední inhibitor CYP3A4, dávka acalabrutinibu by měla být redukována na 100 mg jednou denně. Obdobná, jen opačná situace je se silnými induktory CYP3A4. V této situaci je třeba vyloučit souběžné podávání. Pokud se nelze souběžnému podávání vyhnout, lze zvážit zvýšení dávky acalabrutinibu na 200 mg dvakrát denně [14].
Indikace
Indikace acalabrutinibu se odvíjejí od dosavadních poznatků z klinických studií. Acalabrutinib je indikován v monoterapii nebo v kombinaci s obinutuzumabem k léčbě dospělých pacientů s dříve neléčenou CLL a dále je indikován k léčbě dospělých pacientů s CLL, kteří podstoupili alespoň jednu předchozí léčbu.
Kontraindikace
Známá nebo předpokládaná přecitlivělost na acalabrutinib jako léčivou látku nebo na kteroukoliv pomocnou látku zmíněnou jako obsah tobolky, tvořící tvrdou tobolku anebo inkoust použitý k potisku [14].
Nežádoucí účinky
Acalabrutinib se musí užívat
v souladu s pokyny výrobce. Jak bylo popsáno výše,
dávkování a zacházení s acalabrutinibem by mělo vždy
být v souladu s principem minimalizace rizik. Veškerá
dostupná data týkající se acalabrutinibu vycházejí
z klinických údajů a jsou detailně popsána v tabulce 1.
Přerušení léčby nebo snížení dávky v důsledku nežádoucích účinků
Ve skupině 1 040 pacientů bylo hlášeno přerušení léčby z důvodu výskytu NÚ u 9,3 % osob, přičemž mezi hlavní NÚ patřily pneumonie, trombocytopenie a průjem. Ve skupině 223 pacientů byl výskyt přerušené léčby 10,8 % při stejných reakcích. Snížení dávky kvůli NÚ bylo zaznamenáno u 4,2 % pacientů, přičemž mezi hlavní NÚ patřily reaktivace hepatitidy B, sepse a průjem. Snížení dávky u 223 pacientů z důvodu výskytu NÚ bylo zaznamenáno u 6,7 % osob a mezi hlavní NÚ patřily neutropenie, průjem a zvracení.
V klinických studiích nebyl pozorován žádný klinicky relevantní rozdíl v bezpečnosti a účinnosti mezi pacienty staršími 65 let a mladšími, a to jak v monoterapii, tak v kombinované léčbě [14].
Lékové interakce
Jak bylo popsáno výše, in vitro studie nepotvrdily u obou látek ovlivnění CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6. Látka je metabolizována hlavně CYP3A a v menší míře konjugací s glutationem a hydrolýzou amidů. Metabolismus acalabrutinibu na hlavní aktivní metabolit ACP 5862 je zprostředkován právě tímto izoenzymem.
Acalabrutinib je slabým inhibitorem CYP3A4/5, 2C8 a 2C9 a slabým induktorem izoenzymů CYP3A4, 1A2 a 2B6. ACP 5862 je slabým inhibitorem izoenzymů CYP2C8, 2C9 a 2C19 a slabým induktorem CYP3A4. Acalabrutinib je substrátem P gp a BCRP.
Léčivé látky, které mohou zvyšovat plazmatické koncentrace acalabrutinibu
Souběžné podávání acalabrutinibu se silným inhibitorem CYP3A/P gp (200 mg itrakonazolu jedenkrát denně po dobu pěti dnů) u zdravých subjektů (n = 17) zvýšilo hodnotu cmax a AUC acalabrutinibu 3,9krát, resp. pětkrát. Je třeba vyloučit souběžné užívání přípravku se silnými inhibitory CYP3A/P pg. Jestliže silné inhibitory CYP3A/P gp, jako jsou ketokonazol, konivaptan, klaritromycin, indinavir, itrakonazol, ritonavir, telaprevir, posakonazol, vorikonazol, budou užívány krátkodobě, je třeba přerušit léčbu acalabrutinibem.
Léčivé látky, které mohou snižovat plazmatické koncentrace acalabrutinibu
Souběžné podávání silného induktoru CYP3A (600 mg rifampicinu jednou denně po dobu devíti dnů) u zdravých subjektů (n = 24) snížilo cmax a AUC acalabrutinibu o 68 %, resp. 77 %. Obecně se doporučuje vyloučit souběžné podávání acalabrutinibu se silnými induktory CYP3A (např. fenytoin, rifampicin, karbamazepin). Je potřeba vyloučit souběžné podávání s třezalkou tečkovanou, která navíc může nepředvídatelně snižovat plazmatické koncentrace acalabrutinibu.
Léčivé přípravky snižující množství žaludeční kyseliny
Rozpustnost acalabrutinibu klesá se zvyšujícím se pH. Souběžné podávání acalabrutinibu s 1 g uhličitanu vápenatého u zdravých subjektů snížilo AUC acalabrutinibu o 53 %. Souběžné podávání s 40 mg omeprazolu po dobu pěti dnů snížilo AUC acalabrutinibu o 43 %.
Je li vyžadována léčba látkou snižující množství žaludeční kyseliny, je třeba zvážit použití antacida (např. uhličitanu vápenatého) nebo antagonisty H2 receptorů (např. ranitidin nebo famotidin). V případě užívání s antacidy musí být dodržen odstup mezi podáním obou přípravků a acalabrutinib je třeba užít alespoň dvě hodiny před podáním antagonisty H2 receptoru nebo 10 hodin po něm. Vzhledem k dlouhodobému účinku inhibitorů protonové pumpy by časový odstup dávek interakci neminimalizoval, a proto je třeba souběžné užívání vyloučit.
Léčivé látky, jejichž plazmatické koncentrace mohou být ovlivněny acalabrutinibem
In vitro údaje naznačují, že acalabrutinib působí jako inhibitor CYP3A4 na úrovni střevní sliznice a může zvyšovat expozici substrátům CYP3A4 citlivým na střevní metabolismus. Doporučena je opatrnost při podávání acalabrutinibu se substráty CYP3A4 s úzkým terapeutickým rozmezím podávanými per os (např. cyklosporin, ergotamin, pimozid). Další studie in vitro ukazují, že acalabrutinib indukuje CYP1A2. Souběžné podávání acalabrutinibu se substráty CYP1A2 (např. teofylin, kofein) může vést ke snížené expozici.
Účinky na transportní systémy
Acalabrutinib může zvyšovat expozici souběžně podávaným substrátům BCRP (např. metotrexát) inhibicí střevního systému. Pro minimalizaci potenciálu interakce v gastrointestinálním traktu musejí být substráty BCRP s úzkým terapeutickým intervalem užívány per os alespoň šest hodin před podáním acalabrutinibu nebo po něm. ACP 5862 může zvyšovat expozici substrátům MATE1 (např. metformin) prostřednictvím inhibice tohoto systému. Pacienti užívající přípravky, jejichž eliminace závisí na MATE1, musejí být sledováni kvůli změnám snášenlivosti způsobeným zvýšenou expozicí medikaci při podávání acalabrutinibu [14,15].
Upozornění
Těhotenství a laktace
Ženám ve fertilním věku by podávání acalabrutinibu nemělo být doporučeno. O použití acalabrutinibu u těhotných žen nejsou k dispozici žádné údaje. Dostupná jsou pouze in vivo data o existenci rizika expozice plodu acalabrutinibu během těhotenství, kde byla u zvířat pozorována dystokie a při podávání u březích králíků byl pozorován zpomalený růst plodu.
Ze stejného důvodu není acalabrutinib doporučen ani pro kojící ženy, jelikož riziko pro kojené dítě nelze vyloučit. Doporučeno je nekojit během léčby a dva dny po užití poslední dávky.
Infekce
U pacientů léčených acalabrutinibem v monoterapii a v kombinaci se objevily závažné infekce bakteriálního, virového nebo mykotického původu včetně fatálních případů. Objevily se infekce způsobené reaktivací viru hepatitidy B a viru herpes zoster, aspergilóza a progresivní multifokální leukoencefalopatie.
Cytopenie
Při léčbě byly pozorovány případy s cytopenií stupně 3 nebo 4, včetně neutropenie, anémie nebo trombocytopenie. Během léčby je třeba sledovat krevní obraz.
Další primární malignity
Případy dalších primárních
malignit včetně kožních a mimokožních karcinomů byly
rovněž hlášeny (v případě kožních karcinomů často).
Pacienty je třeba sledovat z důvodu vzniku kožního karcinomu
a poučit je o ochraně před slunečním zářením.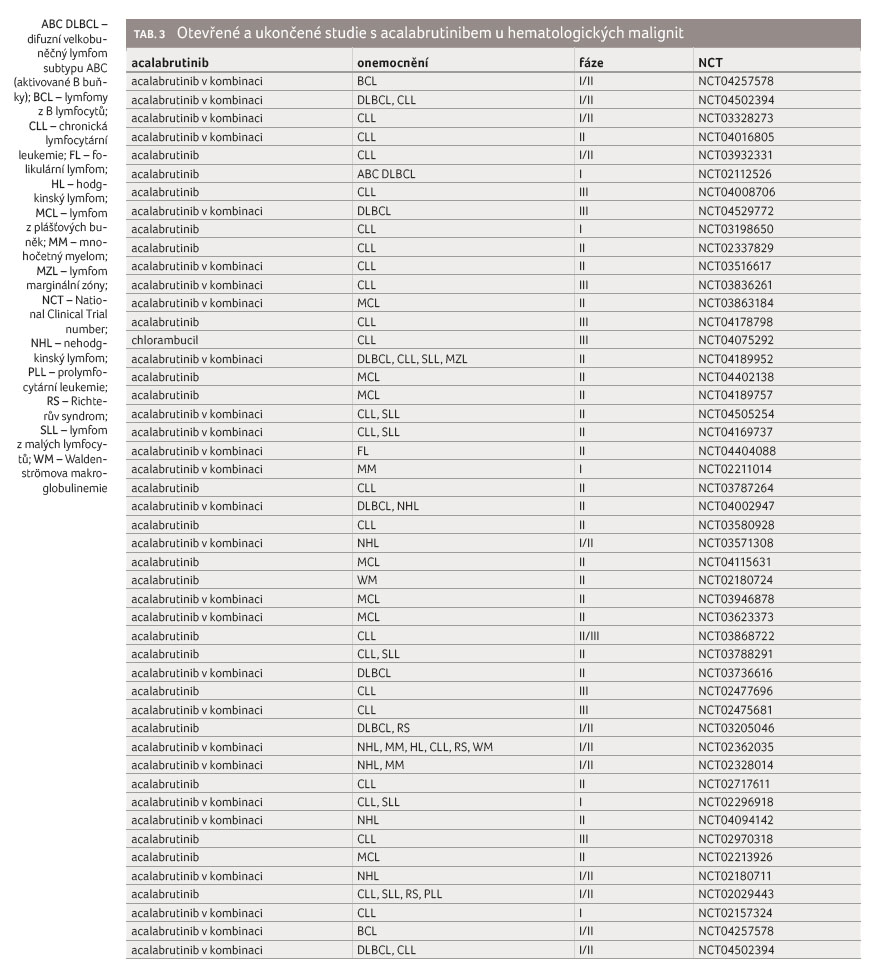
Atriální fibrilace
U pacientů byly také pozorovány atriální fibrilace/flutter. Nemocné je třeba sledovat kvůli symptomům (např. palpitace, závrať, synkopa, bolest na hrudi, dušnost) atriální fibrilace a atriálního flutteru a zajistit vyšetření EKG. Zároveň je při navození atriální fibrilace namístě důkladné zhodnocení rizika tromboembolické nemoci.
Krvácení
Zároveň byly pozorovány závažné
krvácivé příhody včetně krvácení do centrálního
nervového systému a gastrointestinálního krvácení, některé
s fatálním následkem. Celkově byly krvácivé příhody méně
závažné. Mechanismus vzniku krvácivých příhod není zcela znám
[14].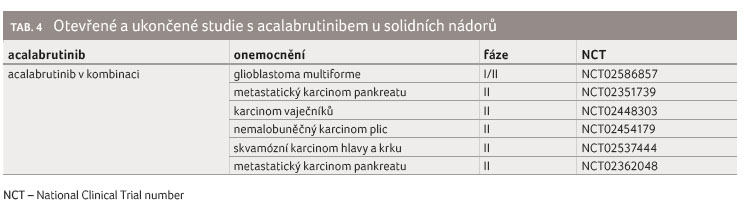
Závěr
Acalabrutinib je vysoce selektivní ireverzibilní inhibitor BTK druhé generace. Látka má prokázanou lepší cílovou specificitu a zvýšenou účinnost pro BTK, což vede k méně NÚ a k nižší toxicitě. Aktuálně probíhá celá řada klinických studií hodnotících léčivo u dalších onkologických onemocnění (tab. 3 a 4) v monoterapii nebo v kombinacích s jinými látkami (nové protilátky proti CD19 a CD20, inhibitory BCL 2, inhibitory PI3 kinázy, inhibitory ALK atd.) [16].
Postupem času bude zajímavé sledovat, zda se mechanismy rezistence budou lišit od mechanismů pro ibrutinib. V aktivním klinickém vývoji se aktuálně nacházejí další selektivní inhibitory BTK, tj. ONO/GS 4059 a BGB 3111 [17].
Seznam použité literatury
- [1] Chronic Lymphocytic Leukemia Treatment (PDQ®) – Health Professional Version ‒ National Cancer Institute. Comprehensive Cancer Information ‒ National Cancer Institute [online]. Dostupné na: https://www.cancer.gov/types/leukemia/hp/cll‑treatment‑pdq
- [2] Doubek M, Špaček M, Pospíšilová Š, et al. Doporučení pro diagnostiku a léčbu chronické lymfocytární leukemie (CLL) – 2018. Transfuze Hematol dnes 2018; 24: 208–220.
- [3] Covey T, Barf T, Gulrajani M, et al. Abstract 2596: ACP‑196: a novel covalent Bruton’s tyrosine kinase (Btk) inhibitor with improved selectivity and in vivo target coverage in chronic lymphocytic leukemia (CLL) patients. Cancer Res 2015; 75(15 Suppl): 2596.
- [4] Byrd JC, Harrington B, O′Brien S, et al. Acalabrutinib (ACP‑196) in relapsed chronic lymphocytic leukemia. N Engl J Med 2016; 374: 323–332.
- [5] Gardner HL, Harrington BK, Izumi R, et al. Abstract 1744: ACP‑196: a second generation Btk inhibitor demonstrates biologic activity in a canine model of B‑cell non‑Hodgkin lymphoma. Cancer Res 2014; 74(19 Suppl): 1744.
- [6] Niemann CU, Montraveta A, Herman SEM, et al. Abstract 2624: the novel Bruton’s tyrosine kinase inhibitor ACP‑196 shows in vivo efficacy against human chronic lymphocytic leukemia cells xenografted to the NSG mouse model. Cancer Res 2014; 74(19 Suppl): 2624.
- [7] Herman SEM, Montraveta A, Niemann CU, et al. The Bruton tyrosine kinase (BTK) inhibitor ACP‑196 demonstrates clinical activity in two mouse models of chronic lymphocytic leukemia. Blood 2015; 126: 2920.
- [8] Sharman JP, Egyed M, Jurczak W, et al. Acalabrutinib with or without obinutuzumab versus chlorambucil and obinutuzmab for treatment‑naive chronic lymphocytic leukaemia (ELEVATE TN): a randomised, controlled, Phase 3 trial. Lancet 2020; 395: 1278‒1291.
- [9] Ghia P, Pluta A, Wach M, et al. ASCEND: Phase III, Randomized Trial of Acalabrutinib Versus Idelalisib Plus Rituximab or Bendamustine Plus Rituximab in Relapsed or Refractory Chronic Lymphocytic Leukemia. J Clin Oncol 2020; 38: 2849‒2861.
- [10] Furman RR, Byrd JC, Own RG, et al. Safety of acalabrutinib (acala) monotherapy in hematologic malignancies: pooled analysis from clinical trials. J Clin Oncol 2020; 38(Suppl 15); Abstr 8064.
- [11] Byrd JC, Wierda WG, Schuh A, et al. Acalabrutinib monotherapy in patients with relapsed/refractory chronic lymphocytic leukemia: updated Phase 2 results. Blood 2020; 135: 1204‒1213.
- [12] Bond DA, Alinari L, Maddocks K. Bruton tyrosine kinase inhibitors for the treatment of mantle cell lymphoma: review of current evidence and future directions. Clin Adv Hematol Oncol 2019; 17: 223‒233.
- [13] Wu J, Zhang M, Liu D. Acalabrutinib (ACP‑196): a selective second‑generation BTK inhibitor. J Hematol Oncol 2016; 9; doi.org/10.1186/s13045‑016‑0250‑9.
- [14] SPC Calquence (acalabrutinib). Státní ústav pro kontrolu léčiv, 2021. Dostupné na: https://www.sukl.cz/modules/medication/detail.php? code=0249959&tab=texts [navštíveno 20. 1. 2021]
- [15] Zhou D, Podoll T, Xu Y, et al. Evaluation of the Drug‑Drug Interaction Potential of Acalabrutinib and Its Active Metabolite, ACP‑5862, Using a Physiologically‑Based Pharmacokinetic Modeling Approach. CPT Pharmacometrics Syst Pharmacol 2019; 8: 489‒499.
- [16] Walter HS, Rule SA, Dyer MJS, et al. A Phase 1 clinical trial of the selective BTK inhibitor ONO/GS‑4059 in relapsed and refractory mature B‑cell malignancies. Blood 2016; 127: 411–419.
- [17] Tam C, Grigg AP, Opat S, et al. The BTK inhibitor, Bgb‑3111, is safe, tolerable, and highly active in patients with relapsed/refractory B‑cell malignancies: initial report of a Phase 1 first‑in‑human trial. Blood 2015; 126: 832.
- [18] National Comprehensive Cancer Network. Chronic Lymphocytic Leukemia/Small Lymfocytic Lymphoma (version 1.2021). Dostupné na: http://www.nccn.org/professionals/physician_gls/default.aspx [navštíveno leden 2021].
- [19] Eichhorst B, Robak T, Montserrat E, et al. ESMO Guidelines Committee. Chronic lymphocytic leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow‑up. Ann Oncol 2021; 32: 23‒33.