Cemiplimab
Souhrn:
Důra M. Cemiplimab. Remedia 2021; 31: 455–460.
Imunoterapie monoklonálními protilátkami prožívá svůj zlatý věk na poli onkologické léčby. Tato léčba dosahuje signifikantních a trvalých léčebných odpovědí a přináší prodloužení doby do progrese a prodloužení celkového přežití za současného zlepšení kvality života. Cemiplimab je plně humánní monoklonální protilátka, která je v současné době hrazena u dospělých pacientů s lokálně pokročilým nebo metastazujícím spinocelulárním karcinomem kůže, kteří nejsou kandidáty pro kurativní operaci či radioterapii. Článek předkládá lékový profil cemiplimabu se zaměřením na jeho indikační omezení, terapeutický efekt a nežádoucí účinky.
Summary:
Dura M. Cemiplimab. Remedia 2021; 31: 455–460.
Immunotherapy with monoclonal antibodies passes through the golden age in the field of antitumoral therapy. This kind of therapy achieves significant and long‑lasting treatment responses and reveals prolongation of progression free survival, overall survival together with quality‑of‑life improvement. Cemiplimab is a fully human monoclonal antibody which is currently reimbursed in the treatment of adult patients with metastatic or locally advanced cutaneous squamous cell carcinoma who are not candidates for curative surgery or curative radiation. The article discusses the cemiplimab drug profile, focused on its limitation of use, therapeutic effect and adverse events.
Key words: cemiplimab – immunotherapy – squamous cell carcinoma.
Úvod
Spinocelulární karcinom kůže (squamous cell carcinoma, SCC) je po bazocelulárním karcinomu druhým nejčastějším zhoubným kožním nádorem. Biologické chování SCC se odvíjí od přítomnosti jeho rizikových faktorů. Mezi ty patří makroskopická velikost tumoru nad 2 cm, lokalita na spánku, ušním boltci či rtu, vysoký histopatologický grading (tzn. nízká diferenciace), invaze do koria hloubky více než 6 mm, perineurální šíření, desmoplazie a z celkových rizikových faktorů pak imunosuprese. SCC s těmito vlastnostmi je klasifikován jako vysoce rizikový (high risk), v opačném případě pak nízce rizikový (low risk) [1]. Dle jeho rizikovosti se liší doporučení pro následné sledování pacientů a plánování případných zobrazovacích vyšetření.
SCC se může chovat lokálně agresivně v místě svého vzniku či může zakládat metastázy ve spádových lymfatických uzlinách a vzdálených orgánech. Karcinom s takovým biologickým chováním je označován jako pokročilý kožní SCC, který je někdy rozdělován na lokálně pokročilý, lokoregionálně metastazující a vzdáleně metastazující.
Zlatým standardem léčby pokročilého SCC byla onkochirurgie. V případě inoperabilního či metastazujícího nádoru je indikována systémová léčba či radioterapie. V současně době reprezentuje první linii systémové léčby pokročilého SCC imunoterapeutikum cemiplimab [2].
Cemiplimab (ATC skupina L01XC33) je plně humánní monoklonální protilátka namířená proti receptoru programované buněčné smrti 1 (programmed cell death protein 1, PD 1). Průběh léčebné odpovědi a spektrum nežádoucích účinků cemiplimabu jsou analogické jiným imunoterapeutikům ze skupiny anti PD 1 protilátek.
Informace dále uvedené jsou zčásti čerpány z webových stránek Státního ústavu pro kontrolu léčiv (SÚKL) [3] a z aktuálního Souhrnu údajů o přípravku (SPC) cemiplimabu (Libtayo) [4].
Indikace a indikační omezení
Cemiplimab je dle indikačních omezení SÚKL hrazen u dospělých pacientů s lokálně pokročilým nebo metastazujícím spinocelulárním karcinomem kůže, kteří nejsou kandidáty pro kurativní operaci či radioterapii [3].
Cemiplimab je též indikován pro systémovou terapii lokálně pokročilého či metastazujícího bazocelulárního karcinomu u pacientů, u kterých došlo k progresi při terapii inhibitorem signální kaskády Hedgehog (vismodegib) či u těch, kteří tuto terapii netolerovali. Tato indikace však nemá v současné době (informace k září 2021) schválenu úhradu z veřejného zdravotního pojištění. Z tohoto důvodu se v textu zaměřujeme pouze na terapii pokročilého SCC.
Pacient, který je vhodným kandidátem pro terapii cemiplimabem, musí současně splnit následující podmínky:
- má výkonnostní stav 0–1 dle ECOG (Eastern Cooperative Oncology Group),
- nevykazuje přítomnost symptomatických mozkových metastáz, případné mozkové metastázy jsou adekvátně léčeny,
- není dlouhodobě léčen systémovými kortikosteroidy v dávce prednisonu nad 10 mg denně (tj. dexametasonu nad 1,5 mg denně či metylprednisolonu nad 8 mg denně) nebo jinou systémovou imunosupresivní léčbou,
- nemá diagnostikováno závažné aktivní systémové autoimunitní onemocnění s výjimkou diabetes mellitus 1. typu, autoimunitního zánětu štítné žlázy a kožního autoimunitního onemocnění (např. psoriáza, atopický ekzém, alopecia areata či vitiligo),
má přijatelnou funkci ledvin a jater:
- koncentrace kreatininu ≤ 1,5 horní hranice normy (upper limit of normal, ULN),
- koncentrace bilirubinu ≤ 1,5 ULN, u pacientů s Gilbertovým syndromem ≤ 3 ULN,
- koncentrace aspartátaminotransferázy a alaninaminotransferázy ≤ 3 ULN, v případě přítomnosti jaterních metastáz ≤ 5 ULN,
- má uspokojivé hodnoty krevního obrazu:
- koncentrace hemoglobinu 90 g/l,
- počet leukocytů 2,5 109/l,
- počet neutrofilů 1,5 109/l,
- počet trombocytů 100 109/l.
Terapie je dále vyloučena u pacientů po transplantaci solidního orgánu, u nemocných trpících chronickou lymfatickou leukemií, aktivní virovou hepatitidou typu B a C či známou infekcí lidským virem imunodeficience (HIV).
Léčba je hrazena do progrese onemocnění (která je verifikována opakovaným CT vyšetřením v odstupu 4–8 týdnů z důvodu vyloučení tzv. pseudoprogrese) a/nebo do neakceptovatelné toxicity, maximálně však po dobu dvou let. Při klinicky viditelném postižení je doporučována pravidelná fotodokumentace.
Forma léku a dávkování
Cemiplimab je dodáván jako sterilní koncentrát pro infuzní roztok obsahující 350 mg cemiplimabu v injekční lahvičce o objemu 7 ml (50 mg/ml) [4]. Aplikuje se v plošné dávce 350 mg v infuzi trvající 30 minut každé tři týdny. Ředění léku je možno provádět do fyziologického roztoku či 5% roztoku glukózy.
Naředěný roztok by měl být podán bez odkladu. V případě opožděného podání může být skladován při pokojové teplotě do 25 °C po dobu maximálně 8 hodin nebo v chladničce při teplotě 2-8 °C po dobu maximálně 24 hodin.
Premedikace před aplikací cemiplimabu není nutná. Při terapii cemiplimabem je nutná monitorace jaterních a renálních funkcí, krevního obrazu včetně diferenciálního počtu leukocytů, hormonů štítné žlázy a hodnoty ranního kortizolu.
Mechanismus účinku a farmakokinetické vlastnosti
Cemiplimab je plně humánní protilátka typu IgG4, která je namířena proti receptoru PD 1. Cemiplimab je vyráběn technologií rekombinantní DNA v suspenzní kultuře ovariálních buněk křečíka čínského.
Receptor PD 1 je exprimován na T
lymfocytech. Za normálních podmínek dochází při navázání
receptoru PD 1 na jeho ligandy PD L1 či PD L2,
exprimované na antigen prezentujících i nádorových
buňkách, k inhibici funkce T lymfocytu. Při blokaci
receptoru PD 1 cemiplimabem je tento inhibiční signál
přerušen a dochází tím k posílení aktivity
imunitního systému závislé na T lymfocytech (obr. 1).
Posilována je jejich proliferace, sekrece cytokinů a cytotoxická
aktivita.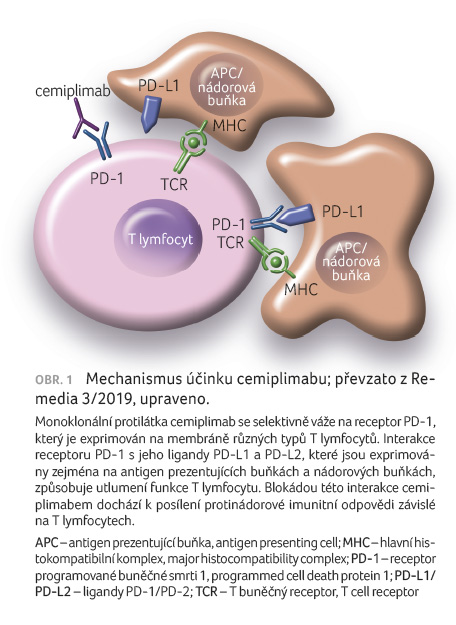
Z klinických hodnocení vyplynulo, že při dávkování cemiplimabu od 1 mg/kg do 10 mg/kg každé dva týdny se jeho kinetika chová lineárně. Ustáleného stavu je dosaženo v průměru za čtyři měsíce. Clearance cemiplimabu po první dávce je v průměru 0,33 l/den. Celková clearance klesá v čase přibližně o 35 % na hodnotu 0,21 l/den, tento pokles se však nezdá být klinicky signifikantní. Biologický poločas eliminace cemiplimabu v době ustáleného stavu činí 19,4 dne. Klinicky signifikantní efekt na farmakokinetické parametry nebyl prokázán u žádného z těchto faktorů – věk, pohlaví, tělesná hmotnost, rasa, typ nádoru, hodnota sérového albuminu, mírná jaterní insuficience a mírná renální insuficience.
Klinická hodnocení a bezpečnost
Terapeutický efekt a bezpečnost cemiplimabu u pacientů s metastazujícím a lokálně pokročilým kožním SCC, kteří nebyli kandidáty na kurativní operační řešení či radioterapii, byl hodnocen v klinickém sledování R2810 ONC 1540 [5]. Jednalo se o otevřenou multicentrickou studii fáze II, do níž bylo zařazeno 193 pacientů s metastazujícím (115 pacientů) a lokálně pokročilým kožním SCC (78 pacientů). Průměrný věk pacientů činil 72 let (38-96 let), výrazně převažovali muži (83,4 %).
Pacienti byli rozděleni do tří skupin – nemocní s metastazujícím SCC s dávkováním 3 mg/kg každé dva týdny (Q2W); nemocní s lokálně pokročilým SCC s týmž dávkováním; nemocní s metastazujícím SCC s plošným dávkováním 350 mg každé tři týdny (Q3W).
Léčba cemiplimabem pokračovala do progrese onemocnění, nezvladatelné toxicity či do dokončení dávkovacího schématu (3 mg/kg Q2W po dobu 96 týdnů; 350 mg Q3W po dobu 54 týdnů).
Výsledným primárním cílovým
ukazatelem byla celková četnost odpovědí, klíčovým sekundárním
ukazatelem byla doba trvání léčebné odpovědi. Stran předchozí
terapie SCC 33,7 % pacientů obdrželo minimálně jednu
protinádorovou systémovou léčbu, 90,2 % pacientů bylo před
vstupem do studie léčeno chirurgicky a 67,9 % pacientů
absolvovalo předchozí radioterapii. Mezi pacienty s metastazujícím
SCC mělo 76,5 % nemocných vzdálené metastázy a 22,6 %
pouze uzlinové metastázy.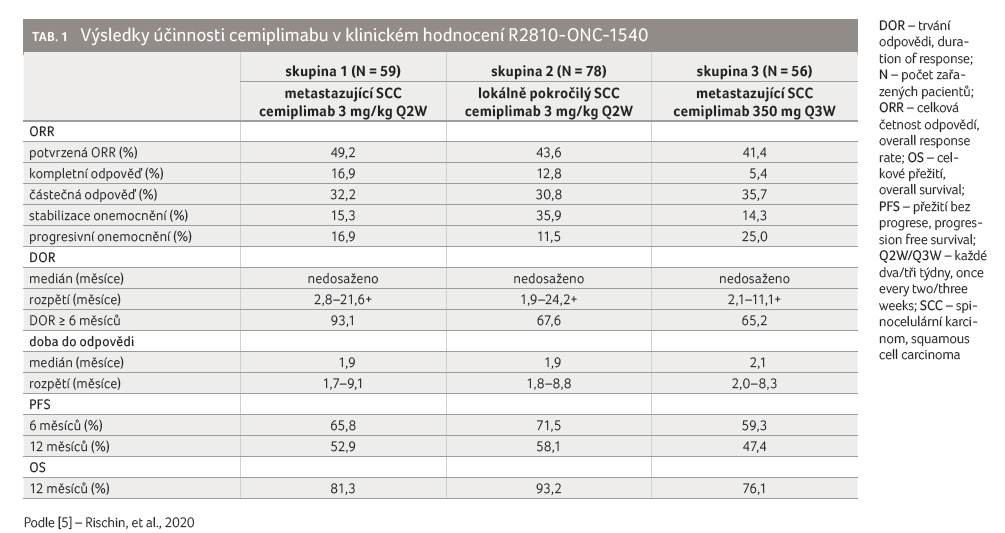
Výsledky klinického hodnocení prezentuje tabulka 1. U všech tří skupin pacientů bylo dosaženo celkové četnosti odpovědí více než 40 %, nejčastěji se jednalo o částečnou odpověď. Trvání odpovědi ≥ 6 měsíců vykázalo napříč všemi skupinami více než 65 % pacientů. Doba do nástupu terapeutické odpovědi se pohybovala kolem dvou měsíců.
Nežádoucí účinky
Četnost a závažnost nežádoucích účinků byla hodnocena v nekontrolovaných klinických studiích u 591 pacientů.
Nežádoucí účinky cemiplimabu jsou imunitně zprostředkované (immune related Adverse Events, irAEs) a v zásadě se neliší od nežádoucích účinků jiných inhibitorů receptoru PD 1. Jejich grading a management jsou hodnoceny na základě kritérií CTCAEv5.0 (Common Terminology Criteria for Adverse Events, verze 5.0) z roku 2017. Hodnocení je pětistupňové, přičemž stupeň 5 značí fatální nežádoucí účinek.
Redukce dávky cemiplimabu se při
výskytu nežádoucího účinku nedoporučuje. Uplatňuje se
přerušení léčby či úplné vysazení léku v závislosti
na závažnosti nežádoucí příhody. Každý pacient by měl
být vybaven příručkou a kartičkou pacienta s názvem
léku a nejčastějšími imunitně zprostředkovanými
nežádoucími účinky, kartičku by měl nosit stále u sebe
pro případ neočekávané potřeby.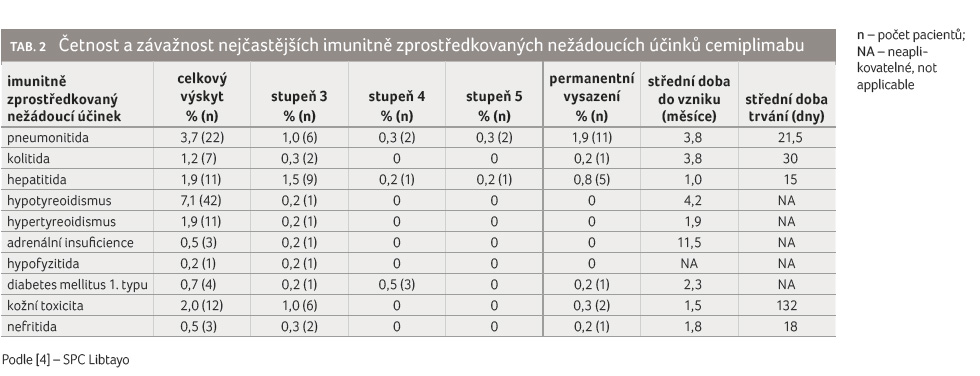
Četnost a závažnost nejčastějších imunitně zprostředkovaných nežádoucích účinků cemiplimabu shrnuje tabulka 2.
Obdobně jako u dalších monoklonálních protilátek namířených proti PD 1 byly nejčastějšími irAEs plicní toxicita ve formě pneumonitidy, gastrointestinální toxicita ve formě kolitidy, hepatotoxicita, endokrinní toxicita (hypo či hypertyreoidismus, adrenální insuficience, hypofyzitida a diabetes mellitus 1. typu), kožní toxicita a renální toxicita ve formě nefritidy.
Z hlediska četnosti byl u cemiplimabu nejčastěji identifikován hypotyreoidismus (v 7,1 %), na druhém místě pak pneumonitida (u 3,7 %). Nejčasnějšími irAEs byly hepatitida se střední dobou vzniku do jednoho měsíce, na druhém místě pak kožní toxicita se střední dobou do vzniku 1,5 měsíce.
Další imunitně zprostředkované nežádoucí účinky, které se v klinických hodnoceních vyskytovaly s frekvencí nižší než 1 %, se týkaly následujících orgánových systémů:
- CNS (meningitida, encefalitida, myasthenia gravis, paraneoplastická encefalomyelitida, Guillainův-Barrého syndrom, chronická zánětlivá demyelinizační polyradikuloneuropatie) a periferní nervový systém (periferní neuropatie),
- kardiovaskulární systém (myokarditida, perikarditida, vaskulitida),
- imunitní systém (imunitní trombocytopenická purpura),
- pohybový systém (artralgie, artritida, svalová slabost, myalgie, polymyalgia rheumatica),
- oční systém (keratitida).
Popsány byly případy reakcí spojených s podáním infuze. V klinickém hodnocení se objevily u 54 (9,1 %) pacientů z celkového počtu 591, z nichž jeden pacient vykázal závažnost stupně 3 a u dvou pacientů byla z tohoto důvodu léčba ukončena. Nejčastějšími symptomy byly pyrexie, nauzea, zvracení, bolest břicha, zimnice a návaly horka.
Přibližně u 1,1 % pacientů léčených cemiplimabem byl hlášen vznik imunogenicity, tedy tvorby protilátek namířených proti cemiplimabu. Neutralizační protilátky však nebyly identifikovány a vznik imunogenicity neovlivňoval farmakokinetické vlastnosti a bezpečnostní profil cemiplimabu.
Interakce
Klinicky významné interakce s jinými, běžně užívanými léky nejsou známy, a to vzhledem k tomu, že cemiplimab není metabolizován pomocí cytochromu P450 jako většina obvyklých xenobiotik. Výjimku tvoří systémové kortikosteroidy, které v dávce vyšší než 10 mg prednisonu denně či jeho ekvivalentu (viz výše) mohou snížit farmakodynamickou aktivitu cemiplimabu a jeho efekt. Indikována je však systémová kortikosteroidní či jiná systémová imunosupresivní terapie v průběhu terapie cemiplimabem ke zvládání nežádoucích účinků.
Kontraindikace
Kontraindikací je pouze hypersenzitivita na účinnou látku či na kteroukoliv z pomocných látek léčivého přípravku, konkrétně na L histidin, sacharózu, L prolin a polysorbát 80.
Těhotenství a kojení
Ženy v produktivním věku by měly užívat účinnou antikoncepci po celou dobu terapie cemiplimabem a ještě čtyři měsíce po poslední dávce léčby.
Nejsou dosud známy zkušenosti s cemiplimabem užívaným v době těhotenství. Zvířecí reprodukční studie s cemiplimabem nebyly provedeny. Ze zvířecích modelů je známo, že inhibice signální cesty PD 1/PD L1 může způsobit imunitně zprostředkované potracení plodu. Lidské protilátky IgG4 (kterou je i cemiplimab) procházejí placentární bariérou, proto se předpokládá vliv na plod. U těhotných žen se terapie cemiplimabem nedoporučuje.
Není známo, zda cemiplimab přechází do mateřského mléka. Vzhledem k tomu, že protilátky IgG4 do mateřského mléka přecházejí, riziko pro dítě nelze vyloučit. Kojení v době terapie cemiplimabem a čtyři měsíce po ukončení léčby se nedoporučuje.
Závěr
Cemiplimab je moderní imunoterapeutikum, které je určeno pro systémovou léčbu inoperabilního, lokálně pokročilého a metastazujícího spinocelulárního karcinomu kůže a jehož princip je založen na blokování receptoru PD 1. Díky svému prokázanému protinádorovému efektu a dobrému bezpečnostnímu profilu v současné době figuruje cemiplimab jakožto léčba první linie.
V blízké budoucnosti můžeme očekávat rozšíření úhrady léčby cemiplimabem z veřejného zdravotního pojištění i pro lokálně pokročilý či metastazující bazocelulární karcinom po selhání terapie inhibitorem signální kaskády Hedgehog a pro nemalobuněčný karcinom plic exprimující PD L1 ve ≥ 50 % nádorových buněk bez aberace genů EGFR, ALK a ROS1. Aktuální informace jsou uvedeny na webových stránkách SÚKL.
Seznam použité literatury
- [1] Stratigos AJ, Garbe C, Dessinioti C, et al.; European Dermatology Forum (EDF), the European Association of Dermato‑Oncology (EADO) and the European Organization for Research and Treatment of Cancer (EORTC). European interdisciplinary guideline on invasive squamous cell carcinoma of the skin: Part 1. Epidemiology, diagnostics and prevention. Eur J Cancer 2020; 128: 60−82.
- [2] Stratigos AJ, Garbe C, Dessinioti C, et al.; European Dermatology Forum (EDF), the European Association of Dermato‑Oncology (EADO) and the European Organization for Research and Treatment of Cancer (EORTC). European interdisciplinary guideline on invasive squamous cell carcinoma of the skin: Part 2. Treatment. Eur J Cancer 2020; 128: 83−102.
- [3] Aktuální informace přístupné z webové stránky www.sukl.cz.
- [4] SPC Libtayo (poslední aktualizace 20. 5. 2020). Dostupné na: https://www.ema.europa.eu/en/documents/product‑information/libtayo‑epar‑product‑information_cs.pdf
- [5] Rischin D, Migden MR, Lim AM, et al. Phase 2 study of cemiplimab in patients with metastatic cutaneous squamous cell carcinoma: primary analysis of fixed‑dosing, long‑term outcome of weight‑based dosing. J Immunother Cancer 2020; 8: e000775.