Cilostazol
Léčba ischemické choroby dolních končetin má dva aspekty – jeden je cílen na zlepšení prognózy (intervence rizikových faktorů aterotrombózy), druhý na zlepšení kvality života, zejména zlepšení tolerance chůze. Právě v této druhé strategii má cilostazol významné postavení.
Mechanismus účinku je dvojí: inhibicí izoenzymu fosfodiesterázy 3A zpomaluje biodegradaci cyklických nukleotidů (cAMP a cGMP). Zvýšení nabídky těchto „druhých poslů“ vede v hladké svalovině cévní stěny k vazodilataci, v trombocytu ke stabilizaci a snížené odpovědi na aktivační podněty. Paralelně cilostazol inhibuje transportér ENT-1 facilitující transmembranózní přesun adenosinu. Tím zvyšuje jeho nabídku a stimuluje purinergní receptory v tkáních. Tato aktivita vede ke zvýšení nabídky cAMP v cévní stěně a v trombocytu a potencuje efekt inhibice fosfodiesterázy.
Indikací cilostazolu je symptomatická léčba nemocných ve stadiu klaudikací. Doloženo je významné prodloužení bezbolestného i maximálního klaudikačního intervalu. Tolerance cilostazolu je dobrá, častější byly pouze nežádoucí účinky vlastní všem vazodilatanciím – bolesti hlavy, palpitace a perimaleolární otoky. Bezpečnost léčby byla výborná, nebyl pozorován zvýšený výskyt krvácení.
V souhlase s těmito vlastnostmi je i hodnocení posledními doporučenými postupy TASC II, kde je uvedeno, že cilostazol je pro klaudikující nemocné lékem s největším prokázaným přínosem.
Cilostazol je významným příspěvkem k farmakoterapii ischemické choroby dolních končetin (ICHDK) ve stadiu klaudikací. V řadě dalších indikací (zejména protidestičkové a antiarytmické působení) je efekt prověřován v rámci klinického hodnocení. I když byl cilostazol syntetizován v Japonsku počátkem osmdesátých let a koncem téhož desetiletí zde byl zaveden do klinické praxe, do České republiky se dostává se zpožděním. Nicméně, jak bude ukázáno v tomto přehledu, jeho terapeutický potenciál v léčbě klaudikací je významný a v dohledné budoucnosti se dá předpokládat jeho využití rovněž v rámci inhibice primární hemostázy. Další potenciál se rýsuje v sekundární prevenci cévních mozkových příhod.
Farmakologická skupina
Inhibitor fosfodiesterázy 3A (PDE-3A), inhibitor zpětného vychytávání adenosinu (inhibitor ekvilibračního transportéru nukleosidů ENT-1), vazodilatans s přídatným protidestičkovým účinkem. Cilostazol náleží do farmakoterapeutické skupiny Antitrombotika, inhibitory agregace krevních destiček kromě heparinu (ATC kód: B01AC23).
Chemické a fyzikální vlastnosti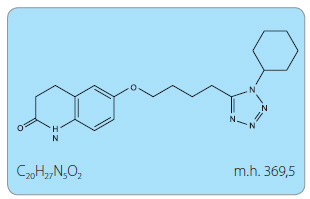
Cilostazol je po chemické stránce derivát chinolinonu, konkrétně (6-[4-(1-cyclohexyl-1H-tetrazol-5-yl)butoxy]-3,4-dihydro-2(1H)-chinolinon (obr. 1). V krystalické či práškové formě má bílou barvu. Po stránce rozpustnosti je cilostazol výrazně lipofilní, mírně rozpustný v ethanolu a prakticky nerozpustný ve vodě.
Sumární vzorec: C20H27N5O2
Molekulová hmotnost: 369,5
Mechanismus účinku
Inhibice izoenzymu fosfodiesterázy 3A
Hlavním účinkem cilostazolu je inhibice PDE-3A v hladké svalovině cévní stěny a v trombocytu. Při terapeutických plazmatických i tkáňových koncentracích je inhibice PDE-3A selektivní, aktivita ostatních izoenzymů fosfodiesterázy (PDE) není prakticky ovlivněna [1]. Hodnota inhibiční konstanty C50 pro PDE-3A je 0,2 μM.
Rodina PDE je souborem 12 izoenzymů regulujících nabídku „druhých poslů“ – cyklického adenosinmonofosfátu (cAMP) či cyklického guanosinmonofosfátu (cGMP) tím, že katalyzují jejich biodegradaci hydrolýzou. Výsledkem inhibice PDE je zvýšení nabídky těchto cyklických nukleotidů. Fosfodiesterázy jsou přítomny téměř ve všech tkáních (CNS, myokard, hladká svalovina cév, bronchů, trávicí trubice, plíce, močovody, děloha apod.). Řada léčiv (např. některá anxiolytika, léky erektilní dysfunkce, spasmolytika, bronchodilatancia) je založena na semiselektivní či selektivní inhibici jednotlivých PDE. Rovněž účinek methylxantinů (theofylinu, kofeinu aj.) je dán neselektivní inhibicí řady PDE.
Vlastní PDE-3 má dvě izoformy – 3A a 3B. Izoforma 3A, jejíž aktivita je cilostazolem inhibována, je lokalizována v hladké svalovině cévní stěny, v trombocytech, v myokardu, endotelu a v reprodukčních tkáních. Její funkcí je kontrola cirkulace a fertility. Naopak izoforma 3B, lokalizovaná v adipocytech a hepatocytech, reguluje lipolýzu.
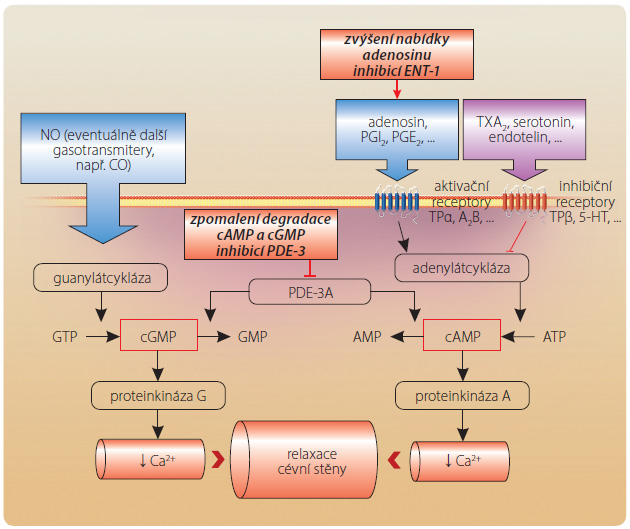
Fosfodiesteráza 3A patří mezi PDE katalyzující hydrolýzu jak cAMP, tak cGMP. I když je afinita k cAMP několikanásobně vyšší, zvýšení nabídky cGMP se uplatní v tkáních s vyšší nabídkou oxidu dusnatého (NO) a guanylátcyklázy, jak je tomu zejména v hladké svalovině cévní stěny. Zde se vazodilatační účinek na podkladě zvýšené nabídky cAMP i cGMP potencuje (obr. 2).
Inhibice ekvilibračního transportéru nukleosidů ENT-1
Druhým, rovněž důležitým účinkem cilostazolu je inhibice zpětného vychytávání (reuptake) adenosinu útlumem ENT-1, což vede ke zvýšení nabídky adenosinu v tkáních, méně již v plazmě. Adenosin aktivuje metabotropní receptory typu G (spřažené s G-proteinem). Ty jsou dvojího typu – aktivační (Gs) a inhibiční (Gi). Receptor typu A2 v cévní stěně či v trombocytu aktivuje adenylátcyklázu a zvyšuje nabídku cAMP. Výsledkem je vazodilatace a stabilizace destičky. Oba pochody – inhibice PDE-3A i inhibice zpětného vstřebávání adenosinu – tak působí v cévách a v trombocytech synergisticky. Naopak receptor typu A1 v myokardu adenylátcyklázu inhibuje a vede ke snížení nabídky cAMP. Působí tak antagonisticky, snížením syntézy cAMP je vlastní efekt inhibice PDE oslaben [2]. Potenciace účinku na cévy a naopak omezení působení na myokard má klinický význam – zvýšena je jak účinnost, tak bezpečnost.
Farmakodynamické působení
Na výsledném farmakodynamickém účinku cilostazolu na úrovni jednotlivých systémů se podílejí oba mechanismy, jak inhibice izoenzymu PDE-3A, tak inhibice transportéru ENT-1.
Účinek na hladkou svalovinu cévní stěny
Inhibice PDE-3A v hladké svalovině cévní stěny (predominantně v končetinovém řečišti, méně již v koronární a mozkové cirkulaci) zvýší nabídku cAMP zpomalením její degradace. Zvýšená nabídka tohoto cyklického nukleotidu aktivuje proteinkinázu A, která fosforylací inhibuje kalciový kanál, nižší koncentrace vápníkových iontů vede ke snížené aktivaci lehkých řetězců myosinu. Výsledkem je relaxace hladké svaloviny a vazodilatace (obr. 2). Celý děj je potencován zvýšením nabídky adenosinu inhibicí jeho reuptake (via ENT-1). Lokální aktivace receptorů A2 v cévní stěně aktivuje adenylátcyklázu (receptory A2 jsou stimulační, typu Gs) s následným zvýšením konverze ATP na cAMP. Vazodilatace je pak navozena souhrou zvýšené syntézy cAMP i zpomalené degradace tohoto „druhého posla“ [3]. Vazoaktivní působení v periferní cirkulaci je základním farmakodynamickým efektem, jehož v praxi využíváme. Větší účinek na konduktivní část tepenného řečiště končetin a menší na ostatní povodí (např. absence účinku na renální arterie) je dán rozdílnou expresí PDE-3A v těchto systémech. Například v koronárním řečišti je vazodilatace kontrolována PDE-1, v plicním řečišti či v kavernózním systému penisu naopak PDE-5.
Účinek na endotel
Obecně lze říci, že cilostazol snižuje negativní dopad ischemie na regulační úlohu endotelu. Tento účinek není plně objasněn a může být vysvětlen řadou mechanismů. Do jaké míry stojí v pozadí snížení rozsahu ischemie, či zda se jedná o primární efekt inhibice PDE-3A, eventuálně ENT-1 (event. o další zatím neobjasněný účinek), není zřejmé.
Experimentálně je doložen efekt cilostazolu na snížení uvolňování hypoxia-inducible factor-1 (HIF-1) [4]. Tato transkripční regulační molekula reguluje metabolismus glycidů v tkáni, diferenciaci, proliferaci a apoptózu buněk. Vzhledem k tomu, že v experimentu na zvířeti byl prokázán ochranný vliv cilostazolu na zhoršení kognitivních funkcí navozené chronickou ischemií a apoptózou buněk, předpokládá se spoluúčast tohoto účinku na příznivém působení u pacientů s ischemickými mozkovými příhodami [4].
Vedle inhibice HIF-1 cilostazol významně stimuluje nabídku řady růstových, vazodilatačních a dalších působků účastnících se na odpovědi tkání na hypoxii či kontrolujících reparační procesy. Na modelu neoangiogeneze navozené ischemií je doložen vliv up-regulace mitogenních molekul, zejména G-CSF (granulocyte colony-stimulating factor) a VEGF (vascular endothelial growth factor), navozené cilostazolem na tvorbu kolaterál [5]. Farmakologická inhibice VEGF efekt cilostazolu eliminovala. Podobně je doložena aktivace či up-regulace některých dalších růstových faktorů a vazoadhezivních molekul. Klinický dopad však zůstává nejasný.
V neposlední řadě se na vazodilataci navozené cilostazolem podílí zvýšení syntézy řady základních vazodilatačních působků – prostacyklinu, prostaglandinu E1 a NO (přehled viz [6]). Vedle zvýšení nabídky se spoluúčastní potenciace výsledné odpovědi na tyto molekuly. Jak prostanoidy, tak NO totiž (cestou aktivace adenylátcyklázy či guanylátcyklázy) zvyšují nabídku cAMP a cGMP. Zpomalení degradace těchto cyklických nukleotidů pak jejich účinek prodlužuje.
Účinek na trombocyty
Obdobně snížení aktivity PDE-3A v trombocytu zvýší nabídku cAMP a stabilizuje destičku (obr. 3). Aktivace či naopak stabilizace trombocytu je řízena po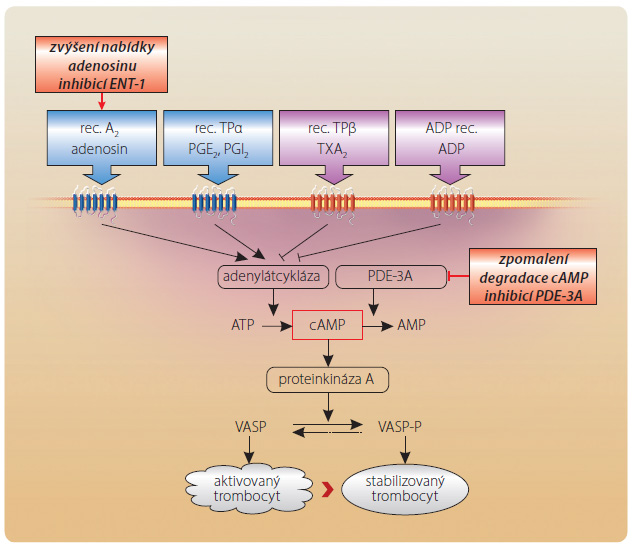 měrem fosforylované a defosforylované formy regulačního proteinu VASP (vasodilator-stimulated phosphoprotein). Zvýšení koncentrace cAMP aktivuje proteinkinázu A, která tento protein fosforyluje a trombocyt stabilizuje. Na zvýšení nabídky cAMP se může účastnit též inhibice transportéru ENT-1. Adenosinové receptory A2 jsou též osazeny na membráně trombocytu a stimulace adenylátcyklázy (obdobně jako v cévní stěně) zvyšuje syntézu cAMP. I tento mechanismus může přispívat k výslednému účinku. Efekt na primární hemostázu je dobře doložen, výsledky klinického hodnocení dokumentují klinický význam, nicméně primární užití cilostazolu v rámci antitrombotické léčby není regulačními orgány schváleno.
měrem fosforylované a defosforylované formy regulačního proteinu VASP (vasodilator-stimulated phosphoprotein). Zvýšení koncentrace cAMP aktivuje proteinkinázu A, která tento protein fosforyluje a trombocyt stabilizuje. Na zvýšení nabídky cAMP se může účastnit též inhibice transportéru ENT-1. Adenosinové receptory A2 jsou též osazeny na membráně trombocytu a stimulace adenylátcyklázy (obdobně jako v cévní stěně) zvyšuje syntézu cAMP. I tento mechanismus může přispívat k výslednému účinku. Efekt na primární hemostázu je dobře doložen, výsledky klinického hodnocení dokumentují klinický význam, nicméně primární užití cilostazolu v rámci antitrombotické léčby není regulačními orgány schváleno.
Účinek na myokard
Situace v myokardu je složitější – zde působí dva mechanismy. Na jedné straně inhibice PDE-3A zpomalí degradaci cAMP a zvýší jeho nabídku v kardiomyocytu (se zvýšením kontraktility, dráždivosti i zvýšením odpovědi k působení katecholaminů). Na straně druhé inhibice transportéru ENT-1 sníží reuptake adenosinu a aktivuje adenosinové receptory A1 v myokardu. Na rozdíl od receptorů A2 v cévách však receptory A1 v myokardu mají inhibiční charakter (jedná se o receptory typ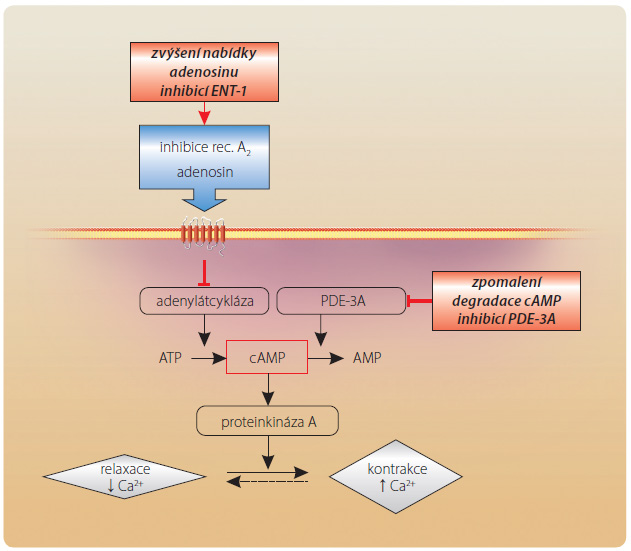 u Gi) a tlumí aktivitu adenylátcyklázy (obr. 4). Snížení syntézy cAMP tak kompenzuje zpomalení degradace tohoto cyklického nukleotidu navozené základním efektem, tedy inhibicí PDE-3A [7]. Prakticky se tak neuplatní farmakodynamický efekt inhibice PDE-3A na myokard, který by vedl k pozitivně chronotropnímu účinku, pozitivně inotropnímu účinku, pozitivně bathmotropnímu účinku (zvýšení excitability) a ke zvýšení citlivosti ke katecholaminům, tedy s efektem, se kterým se setkáváme při podávání ostatních inhibitorů PDE-3, konkrétně milrinonu a amrinonu. Právě ovlivnění zpětného vychytávání adenosinu je hlavní předností cilostazolu, která významně zvyšuje bezpečnost léčby tímto přípravkem [8]. KIinickým korelátem specifického působení cilostazolu je pak podstatně nižší arytmogenní potenciál, či u některých typů arytmií dokonce antiarytmický účinek (např. u Brugadova syndromu).
u Gi) a tlumí aktivitu adenylátcyklázy (obr. 4). Snížení syntézy cAMP tak kompenzuje zpomalení degradace tohoto cyklického nukleotidu navozené základním efektem, tedy inhibicí PDE-3A [7]. Prakticky se tak neuplatní farmakodynamický efekt inhibice PDE-3A na myokard, který by vedl k pozitivně chronotropnímu účinku, pozitivně inotropnímu účinku, pozitivně bathmotropnímu účinku (zvýšení excitability) a ke zvýšení citlivosti ke katecholaminům, tedy s efektem, se kterým se setkáváme při podávání ostatních inhibitorů PDE-3, konkrétně milrinonu a amrinonu. Právě ovlivnění zpětného vychytávání adenosinu je hlavní předností cilostazolu, která významně zvyšuje bezpečnost léčby tímto přípravkem [8]. KIinickým korelátem specifického působení cilostazolu je pak podstatně nižší arytmogenní potenciál, či u některých typů arytmií dokonce antiarytmický účinek (např. u Brugadova syndromu).
Účinek na aterogenezi a na rizikové faktory
V experimentu byl dobře dokumentován antiaterogenní efekt v modelu na zvířeti. Například byla zmenšena plocha aterosklerotického postižení stěny aorty po cholesterolové dietě u myší s deficitem LDL receptoru; paralelně bylo doloženo zlepšení lipidogramu – pokles LDL cholesterolu a vzestup HDL cholesterolu [9]. V humánní medicíně doklad o antiaterogenním působení nemáme, doložen je pouze příznivý účinek na progresi tloušťky intimy-medie v řadě kontrolovaných studií [10].
V pozadí za experimentálně doloženým antiaterogenním účinkem cilostazolu může stát příznivý efekt na lipoproteinové spektrum, konkrétně na zvýšení hodnot HDL cholesterolu a snížení hodnot triglyceridů. V experimentu je doložen příznivý efekt cilostazolu na úpravu dyslipidemie a zpomalení akumulace lipidů v cévní stěně [11]. Průměrné snížení triglyceridemie se pohybovalo kolem 15 %, zvýšení hladiny HDL cholesterolu kolem 10 %. Obdobná úprava dyslipidemie je doložena i v klinických studiích, např. u diabetiků [12].
Farmakokinetické vlastnosti
Po perorální aplikaci se cilostazol dobře absorbuje, biologická dostupnost se při podání nalačno pohybuje kolem 70 %, při podání s tučným jídlem se zvyšuje na 90 % [13]. Vzhledem k mírné afinitě k efluxní pumpě glykoproteinu P je biologická dostupnost jen málo ovlivněna lékovými interakcemi či farmakogenetickými faktory na této úrovni. Hepatální biotransformace izoenzymy CYP3A a CYP2C19 je středně intenzivní, některé metabolity si zachovávají biologickou aktivitu. Pouze 1 % cilostazolu je eliminováno ve formě mateřské látky. Polymorfismus oxidáz CYP2C5 a CYP2C19 má mírný vliv na expozici cilostazolu, interindividuální variabilita je malá – kolem 40–60 % [14]. Lékové interakce (indukce či inhibice) se projevují na expozici mateřské látce málo významně, např. velmi silný inhibitor CYP3A4 ketokonazol zvýšil expozici asi dvojnásobně: snížení koncentrace aktivního metabolitu pak kompenzovalo zvýšení koncentrace mateřské látky.
Daleko větší význam mají změny v koncentraci aktivních metabolitů. Oxidázy CYP3A (zejména 3A4) katalyzují transformaci na dehydrometabolity, naopak CYP2C19 na trans-hydroxymetabolity [15]. Dehydrometabolity jsou přitom několikanásobně (4–7krát) účinnější než mateřská látka (měřeno antiagregační aktivitou). Farmakologická aktivita trans-hydroxymetabolitů je na druhé straně velmi malá (asi pětinová). Inhibice oxidázy CYP3A4, která se projeví zvýšenou tvorbou neaktivních trans-metabolitů, má menší klinický význam. Mírné, maximálně dvojnásobné zvýšení expozice mateřskému cilostazolu je vyrovnáno sníženou transformací na aktivní metabolity. Induktory CYP3A4 sice sníží hladinu cilostazolu, ale zvýší přeměnu na aktivnější dehydrometabolity, i zde nebude mít interakce velký význam. K silným inhibitorům CYP3A4 patří zejména ketokonazol, flukonazol, klarithromycin a některá antiretrovirotika; k induktorům náleží řada antikonvulziv, rifampicin či třezalka.
Na straně druhé, inhibice izoenzymu 2C19 (např. inhibitory protonové pumpy) potencuje transformaci na aktivnější dehydrometabolity a farmakodynamický účinek (měřený protidestičkovým efektem) významně zvýší. Např. současná aplikace omeprazolu zvýšila expozici cilostazolu pouze o pětinu, expozice i koncentrace aktivního dehydrometabolitu však stouply o dvě třetiny. Zde je redukce dávky cilostazolu na polovinu zcela namístě [16].
Zda se efekt aktivních metabolitů cilostazolu projeví pouze v akcentaci protidestičkového účinku, či zda se projeví i na vazodilataci, není zřejmé. Díky v ětší hydrofilii aktivního metabolitu se může zvýšit koncentrace v plazmě, a nikoli v tkáni, potencován tak může být efekt na trombocyty, ale nikoli na hladkou svalovinu cévní stěny.
ětší hydrofilii aktivního metabolitu se může zvýšit koncentrace v plazmě, a nikoli v tkáni, potencován tak může být efekt na trombocyty, ale nikoli na hladkou svalovinu cévní stěny.
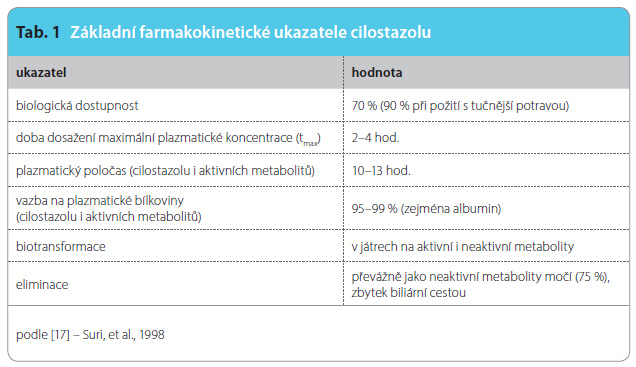
Čas dosažení maximální plazmatické koncentrace při jednorázovém podání 100 mg cilostazolu se pohybuje mezi 2–4 hodinami, (tab. 1) [17]. Plazmatický poločas jak cilostazolu, tak aktivního dehydrometabolitu se pohybuje mezi 10–13 hodinami. Vazba cilostazolu na plazmatické proteiny je vysoká, dosahuje kolem 98 %. Cilostazol i aktivní dehydrometabolit se vylučují po další transformaci ze tří čtvrtin do moče, zbytek je z těla vyloučen biliární cestou [18]. Paradoxem je, že renální selhání mírně snížilo jak expozici cilostazolu, tak vrcholové plazmatické koncentrace cilostazolu i metabolitů ve srovnání se zdravými dobrovolníky [19]. Toto zjištění nepodporuje požadavek na snížení dávky cilostazolu při renální nedostatečnosti. Farmakokinetika při selhání jater nebyla studována, mírné hepatopatie kinetiku neovlivnily.
{kind=link}
Nebyly doloženy rozdíly v absorpci, biologické dostupnosti, distribuci, biotransformaci a v eliminaci cilostazolu a jeho metabolitů na podkladě věku, pohlaví či hmotnosti. V rozmezí šesté až osmé věkové dekády nebyl shledán významný rozdíl v plazmatickém poločase a v ostatních farmakokinetických ukazatelích [17].
Klinické zkušenosti
Léčba symptomatické ischemické choroby dolních končetin
Manifestace ICHDK je velmi rozmanitá – od asymptomatického nemocného diagnostikovaného pomocí sníženého ischemického indexu (ankle brachial index, ABI) přes nemocné s námahou indukovanými bolestmi v končetině až po pacienty trpící klidovými bolestmi, nebo dokonce ulceracemi. Intermitentní klaudikace, jako je námahou vyvolaná bolest způsobená ischemií svalu, je nejtypičtějším příznakem choroby. Pouze u minority nemocných se může jevit bolest jako atypická, pak také často vede k opožděné diagnóze choroby.
Základem léčby projevů periferní aterosklerózy je agresivní intervence známých disponujících stavů – tedy kouření, dyslipoproteinemie, arteriální hypertenze a diabetu. Klaudikující nemocní přicházejí kvůli potížím k lékaři a čekají zlepšení nejen své kardiovaskulární prognózy, ale i stavu své mobility. Medikamentózně léčíme i nemocné asymptomatické (dle Fontainovy klasifikace I. stupně) s cílem prevence dalšího rozvoje aterosklerózy a jejích komplikací. Doklady o příznivém působení užívaných léků pro nemocné s ICHDK extrapolujeme z výsledků velkých studií, do kterých byli zařazeni nemocní jak s koronární, tak s cerebrovaskulární a také s periferní aterosklerózou (např. ze studií Heart Protection Study, CAPRIE nebo HOPE).
Některé léky jsou podávány s cílem zlepšit celkovou kardiovaskulární prognózu – zejména protidestičkové léky, statiny a inhibitory ACE (eventuálně telmisartan), jiné k ovlivnění symptomů choroby (zejména jde o prodloužení klaudikačního intervalu). Léky k ovlivnění symptomů jsou označovány jako vazoaktivní nebo vazodilatační. Do této skupiny tradičně řadíme léky jako pentoxifylin, naftidrofuryl a nověji i cilostazol. V České republice nebyl nikdy užíván pro léčbu klaudikací buflomedil nebo propionyl-L-karnitin. Z nefarmakologických postupů má pro ovlivnění klaudikací zásadní roli svalový trénink, zejména pokud jde o pravidelnou, dózovanou zátěž pod dohledem rehabilitačních specialistů. Tato je však velmi omezeně dostupná.
Mechanismus působení cilostazolu, jak byl popsán výše, spojuje zvýšením koncentrace cAMP a adenosinu přímý vazodilatační vliv na stěnu tepny a současně tlumí primární hemostázu, konkrétně aktivaci trombocytů. Na výsledném efektu se může podílet i zásah do syntézy lipoproteinů, který vede k mírnému snížení hladiny triglyceridů a zvýšení hladiny HDL cholesterolu či k ovlivnění funkce endotelu. Tyto vlastnosti se jeví jako ideální pro nemocné se symptomatickou periferní aterosklerózou.
Cilostazol je registrován pro léčbu klaudikací v zemi výrobce originálního léku (v Japonsku) od roku 1988, ve Spojených státech od roku 1999 a v zemích EU od roku 2000. Zkušenosti jsou tedy dlouhodobé. Studie vedoucí k registraci cilostazolu v léčbě nemocných s ICHDK se uskutečnily většinou již v devadesátých letech, některé na začátku tohoto milénia. Ve studiích prověřujících efekt vazoaktivní medikace na zmírnění klaudikačních potíží je obvykle v testu na nakloněné rovině zkoumáno působení léku na ovlivnění buď bezbolestné klaudikační vzdálenosti (pain-free walking distance, PFWD, v některých studiích také označována jako initial walking distance, ICD), nebo maximální klaudikační vzdálenosti (maximal walking distance, MWD), případně obou ukazatelů. Vzájemná porovnatelnost jednotlivých studií je možná pouze tehdy, pokud je srovnáván rozdíl průměrné dosažené vzdálenosti v souboru na začátku a na konci studie, protože metodika testování se v různých studiích často liší (parametry rychlosti běžícího pásu, jeho náklon). V některých studiích je objektivizována míra končetinové perfuze stanovením ABI v klidu a po zátěži. Jindy je sledována též kvalita života pomocí standardizovaných dotazníků.
Porovnání s placebem
Ve třetí fázi klinického hodnocení cilostazolu bylo provedeno 8 multicentrických, randomizovaných, dvojitě slepých, placebem kontrolovaných studií. Tyto studie vedoucí k registraci léku proběhly většinou ve Spojených státech, v Japonsku a ve Velké Británii. V roce 2002 publikoval Thompson metaanalýzu těchto osmi studií: zahrnuly celkem 2702 nemocných v průměrném věku 65 let, s převahou zařazených mužů – 76 % [20]. Nemocní měli vyjádřeny typické rizikové faktory aterogeneze (hypertenze v 60 %, kouření ve 40 %, diabetes ve 26 %). Délka studií se pohybovala od 12 do 24 týdnů. Nejobvykleji byl podáván cilostazol v dávce 100 mg dvakrát denně (ve dvou studiích i v dávce 50 mg dvakrát denně a v jedné ![Graf 1 Srovnání účinku cilostazolu na prodloužení maximální klaudikační vzdálenosti oproti placebu – metaanalýza osmi studií; podle [24] – Bedenis, et al., 2014.](https://www.remedia.cz/photo-a-29989---.jpg) studii v dávce 150 mg 2krát denně). Výsledky šesti studií ukázaly, že v porovnání s placebem vzrostla maximální klaudikační vzdálenost o 41–54 % (graf 1).
studii v dávce 150 mg 2krát denně). Výsledky šesti studií ukázaly, že v porovnání s placebem vzrostla maximální klaudikační vzdálenost o 41–54 % (graf 1).
Změny hodnot ischemického indexu na začátku studie a na jejím konci a stejně tak proměny hodnot ABI těsně po zátěži studoval Mohler [21]. Vyhodnotil 308 nemocných léčených 100 mg cilostazolu dvakrát denně a 303 nemocných léčených 50 mg dvakrát denně a porovnal výsledky s 299 jedinci dostávajícími placebo. Iniciální hodnota ABI mezi skupinami se nelišila (0,63 vs. 0,62 vs. 0,63). Vlivem léčby došlo k nevelkému vzestupu klidového ABI po 24 týdnech léčby u aktivně léčených (o 0,03, respektive 0,04 proti placebu).
V oblasti léčby ICHDK byly publikovány také výsledky léčby nemocných po periferní endovaskulární intervenci, kdy pacienti následně užívali buď pouze protidestičkový lék, nebo protidestičkovou medikaci a současně cilostazol [22]. Ze dvou randomizovaných studií a čtyř retrospektivně vyhodnocených kohort (celkem 1522 nemocných), sledovaných 18–37 měsíců, vyplynulo, že cilostazol snížil četnost restenóz a nutnost reintervencí, prodloužil dobu přežití bez nutnosti amputace. Tento pozitivní efekt by mohl být vysvětlen uváděným antiproliferativním působením cilostazolu. Omezením tohoto pozorování je fakt, že všechny tyto studie byly prováděny v Japonsku a revaskularizovaná populace byla z hlediska tíže ICHDK značně heterogenní – od indikace revaskularizace pro kritickou končetinovou ischemii po indikaci danou klaudikacemi. Většinou šlo o intervenci v oblasti femoropopliteálního segmentu a 68 % nemocných mělo implantovaný stent.
Lze učinit závěr, že řadou studií bylo doloženo významné prodloužení klaudikačního intervalu (bezbolestného i maximálního) řádově o polovinu, dávka cilostazolu 100 mg 2krát denně měla významně větší efekt než dávka poloviční. Index ABI při léčbě stoupl, efekt však byl minimální. U nemocných po perkutánních intervencích z indikace klaudikací i kritické končetinové ischemie byl doložen nižší výskyt restenóz, nutnost dalších intervencí a prodloužení doby přežití do případné amputace končetiny.
Porovnání s ostatní vazoaktivní léčbou
V symptomatické léčbě ICHDK ve stadiu klaudikací je dosud užíván pentoxifylin a naftidrofuryl. Vzájemné srovnání se logicky nabízí.Ve studii porovnávající cilostazol, pentoxifylin a placebo byl nárůst maximální klaudikační vzdálenosti při podávání cilostazolu 54 %, při podávání pentoxifylinu 34 % a při podávání placeba rovněž 34 % [23]. Výsledky dosažené u pacientů užívajících placebo a pentoxifylin se prakticky nelišily. Nežádoucí účinky, které vedly k přerušení léčby, se vyskytly u 13 %, respektive 16 % nemocných léčených cilostazolem (nižší a vyšší dávkou) a byly přítomny i u 9 % nemocných v placebové větvi a u 21 % nemocných ve větvi pentoxifylinové. Komplexnější pohled podává recentní analýza Cochraneova institutu z roku 2014. Do ní bylo zahrnuto 15 dvojitě slepých randomizovaných studií, ve kterých byl cilostazol porovnáván s placebem nebo s jinou vazoaktivní léčbou [24]. Šlo o souhrn 3718 nemocných, kteří byli léčeni v délce 6–26 týdnů. Data ze 7 studií nemohla být do konečné analýzy zahrnuta kvůli své heterogenitě, pro účely metaanalýzy bylo proto použito pouze 8 studií. Výsledky v parametru bezbolestná vzdálenost ukázaly statisticky významný vzestup hodnot při léčbě oběma studovanými dávkami cilostazolu proti placebu (31,4 m vs. 19,9 m) a při léčbě cilostazolem oproti pentoxifylinu. Stejně tak došlo k vzestupu maximální klaudikační vzdálenosti navozené cilostazolem proti placebu (43,1 m vs. 32 m). Studie s měřením ABI nenašly významný rozdíl ve prospěch cilostazolu. Nebylo zjištěno zvýšení nebo snížení celkové mortality v průběhu sledování, nicméně jednalo se vždy o studie relativně krátkého trvání s malým počtem příhod. Kvalita života hodnocená v nevelkém počtu studií se jevila zlepšena při léčbě cilostazolem. Závěrem Cochraneovy analýzy je řečeno, že cilostazol prodlužuje klaudikační interval a že vedlejší účinky jeho působení jsou mírné. Obdobně byl efekt cilostazolu porovnáván s naftidrofurylem a pentoxifylinem [24]. Naftidrofuryl prodloužil klaudikační vzdálenosti o něco více než cilostazol, rozdíl nedosáhl významnosti, oba léky pak byly účinnější než pentoxifylin. Autoři hodnotí efekt naftidrofurylu i cilostazolu jako velmi dobrý a vhodný k léčbě klaudikací.
Ostatní indikace
Paralelně s léčbou klaudikačních potíží byl potenciál cilostazolu prověřován též v sekundární prevenci mozkových příhod. Zde však máme pouze data z klinických hodnocení, není ovšem schváleno jejich užití. Prvou studií v sekundární prevenci iktů je studie CSPS u japonských nemocných [26]. V tomto hodnocení cilostazol redukoval riziko recidivy příhody o 42 % oproti placebu. I v následné studii CSPS II u nemocných s recentním non-embolickým iktem byla prokázána non-inferiorita cilostazolu ve srovnání s kyselinou acetylsalicylovou (rekurence iktů po 29 měsících 2,7 % a 3,7 %) [27]. V čínské studii CASISP sice došlo také ke snížení počtu iktů oproti kyselině acetylsalicylové (3,3 % vs. 5,6 %), nebylo však dosaženo statistické významnosti [28]. Studie s užitím duální protidestičkové léčby, kdy by jednu složku představoval cilostazol, zatím nemáme, podobně nemáme data u indoevropské populace.
Postavení v rámci konzervativní léčby ICHDK
V rámci farmakoterapie ICHDK se řídíme v současnosti zejména doporučením TASC II (Trans Atlantic Inter-Society Consensus Document on Management of Peripheral Arterial Disease) z roku 2011 a doporučeními evropskými (guidelines ESC pro léčbu periferní aterosklerózy) z téhož roku. V dokumentu TASC II je uvedeno, že ve specifické farmakoterapii nemocných s klaudikacemi jsou účinné a prověřené pouze dva léky – cilostazol a naftidrofuryl. Je zde také uvedeno, že cilostazol je pro klaudikující lékem s největší mírou prokázaného přínosu. Čistý klinický zisk podání cilostazolu na parametr maximální klaudikační vzdálenosti kolísá ve srovnání s placebem od 50 m do 70 m. Také kvalita života na základě hodnocení standardizovanými dotazníky (SF-36 a WIQ) je vyšší při medikaci cilostazolem. Zda se definitivně prověří, jestli má tento lék také význam pro snížení rizika restenóz po endovaskulárních nebo chirurgických zákrocích, je otázkou budoucnosti. Stejně tak je předmětem dalšího výzkumu význam jeho protidestičkového potenciálu, zkoumaný v současnosti u asijské populace v sekundární prevenci iktů. Každopádně pleiotropní působení cilostazolu se jeví již nyní jako maximálně výhodné u nemocných s ICHDK na podkladě aterosklerózy.
Bezpečnost a snášenlivost
Vzhledem k mechanismu účinku je teoreticky možno očekávat několik typů nežádoucích účinků: efekt vazodilatace (edémy, bolesti hlavy, palpitace, hypotenze, závratě, návaly), efekt ovlivnění primární hemostázy (krvácení), efekt zvýšení nabídky adenosinu v tkáních (stimulace dechového centra, převodní poruchy, dyspeptické potíže) či obecné alergické reakce.
V případě cilostazolu máme výhodu, že byla provedena rozsáhlá randomizovaná, zaslepená studie CASTLE, která u více než 1400 pacientů s ICHDK ve stadiu klaudikací sledovala snášenlivost a bezpečnost cilostazolu podávaného v obvyklé dávce 2krát 100 mg. Efekt cilostazolu byl porovnáván s placebem, studie trvala 36 měsíců [29]. Při porovnání snášenlivosti, měřené přerušením léčby, nebyl zásadní rozdíl mezi cilostazolem a placebem. V obou větvích ukončilo léčbu stejné procento nemocných, poměr rizik (hazard ratio, HR) pro cilostazol byl 1,06 (95% konfidenční interval – CI: 0,925–1,224; p = 0,39).
Bezpečnost léčby, měřená tzv. tvrdými daty, byla opět srovnatelná. Při hodnocení celkové mortality bylo riziko pro cilostazol 0,94 (95% CI: 0,64–1,39; p = 0,77), pro kardiovaskulární mortalitu pak byl HR 1,05 (95% CI: 0,502–2,210; p = 0,89). Hodnoty byly srovnatelné při hodnocení on-treatment i intention-to-treat. Výskyt sledovaných nežádoucích příhod byl nevýznamně vyšší ve skupině s léčbou cilostazolem, většina z nich byla ve vztahu k vazo![Graf 2 Výskyt sledovaných nežádoucích příhod při léčbě cilostazolem ve srovnání s placebem ve studii CASTLE; podle [29] – Hiatt, et al., 2008.](https://www.remedia.cz/photo-a-29990---.jpg) dilataci (bolesti hlavy, palpitace, perimaleolární otoky) či k aktivaci purinergních receptorů při vyšší nabídce adenosinu. Závažné nežádoucí účinky – srdeční zástava, krvácení – se naopak nevýznamně častěji vyskytovaly ve skupině placebové, graf 2. Výskyt krvácivých příhod nebyl při léčbě cilostazolem častější ani při současném podávání protidestičkových léků či antikoagulancií (3,3 % vs. 4,4 %).
dilataci (bolesti hlavy, palpitace, perimaleolární otoky) či k aktivaci purinergních receptorů při vyšší nabídce adenosinu. Závažné nežádoucí účinky – srdeční zástava, krvácení – se naopak nevýznamně častěji vyskytovaly ve skupině placebové, graf 2. Výskyt krvácivých příhod nebyl při léčbě cilostazolem častější ani při současném podávání protidestičkových léků či antikoagulancií (3,3 % vs. 4,4 %).
U kardiaků se obáváme poklesu zejména diastolického krevního tlaku (snížení koronární perfuze) a vzestupu srdeční frekvence. Ve studii sledující hemodynamické změny po podání cilostazolu nebyl zaznamenán pokles systolického tlaku, diastolický tlak poklesl v průměru nevýznamně, nicméně maximální pozorovaný pokles činil 29 % [30]. Rovněž srdeční frekvence se významně nezvýšila, maximální vzestup činil 13 %. Maximální účinek byl pozorován 6 hodin po podání léku.
V přehledu Evropské lékové agentury EMA (z 22. března 2013) je proveden rozbor nežádoucích účinků při léčbě cilostazolem. Na základě dat z hlášení (v průběhu 6 milionů pacientoroků) a klinických hodnocení bylo konstatováno průměrné 8% riziko krvácení za dobu léčby cilostazolem (pozorované pouze u nemocných užívajících jiná antitrombotika, samotný cilostazol vyšší výskyt krvácení nenavozoval). Nebyl zjištěn vyšší výskyt poruch srdečního rytmu, doloženo bylo 5% riziko palpitací či zvýšení srdeční frekvence. V kontrolovaných klinických studiích byl souhrnně hlášen stejný počet úmrtí (ze všech příčin i z kardiovaskulárních příčin) ve skupině s cilostazolem i s placebem. Souhrnný ukazatel – výskyt vaskulárních příhod či mortality – byl v postmarketingových studiích opět srovnatelný; např. 135 příhod při léčbě cilostazolem a 153 při podávání placeba ve studii CASTLE.
Samotná analýza osmi významných kontrolovaných studií (1400 nemocných léčených cilostazolem a 1000 nemocných užívajících placebo) dokládá vyšší výskyt příhod spojených s vazodilatací, tj. palpitací či zvýšení srdeční frekvence a bolestí hlavy, nebo spojených s aktivací adenosinových receptorů, tj. změna frekvence vyprazdňování [31]. Ostatní nežádoucí účinky byly stejně časté ve skupině s aktivní léčbou i ve skupině s placebem [32].
Shrneme-li, pak nežádoucí účinky cilostazolu nejsou časté, jsou v souladu s mechanismem účinku a zpravidla nejsou závažné. Nebylo doloženo zvýšení výskytu závažných poruch srdečního rytmu při srovnání s placebem. Nicméně při spontánním hlášení Evropské agentuře bylo referováno několik příhod závažné tachykardie (typ neuveden). Bylo proto vydáno následující doporučení: 1) nepodávat cilostazol nemocným s nestabilní anginou pectoris či s infarktem myokardu, 2) nepodávat v odstupu 6 měsíců od perkutánní koronární intervence, 3) nepodávat nemocným s anamnézou závažné tachyarytmie.
Indikace
Cilostazol je indikován k symptomatické léčbě ICHDK, tedy k prodloužení maximální bezbolestné vzdálenosti u nemocných ve stadiu II Fontainovy klasifikace, tj. u nemocných bez kritické končetinové ischemie. Aplikace cilostazolu je součástí komplexní léčby – jak úpravy životosprávy (zejména abstinence kouření a pohybové léčby), tak farmakoterapie cílené na rizikové faktory aterogeneze, tedy na ovlivnění prognózy. Typickou indikací, v souladu s doporučenými postupy, jsou nemocní ve stadiu IIb, u kterých nebylo úpravou životosprávy či dosavadní farmakoterapií dosaženo dostatečného zmírnění klaudikací. Efekt léčby cilostazolem má být po třech měsících zhodnocen s tím, že pokračování léčby je indikováno pouze při pozitivním účinku. Klinický účinek (vazodilatace i protidestičkový účinek) bývá zřetelný již během 1–2 týdnů, plný efekt však nastupuje později.
Kontraindikace
Cilostazol by neměl být podáván u stavů s vyšším rizikem vzniku závažných nežádoucích účinků. To jsou zejména nemocní s vysokým rizikem krvácení na základě predispozice (zejména po hemoragickém iktu, při vředové chorobě, při proliferativní retinopatii či nekontrolované hypertenzi) nebo při současné medikaci významně ovlivňující hemostázu (duální protidestičková léčba či aplikace antikoagulancií). Dále nepodáváme cilostazol nemocným s vysokým rizikem dysrytmií, tj. se srdečním selháním, s akutními koronárními příhodami (až do odstupu 6 měsíců) s anamnézou závažné tachyarytmie, především komorové tachykardie či fibrilace komor, s multifokálními ektopickými rytmy či nemocným s prodlouženým intervalem QTc.
Dále je kontraindikováno podávání cilostazolu během těhotenství a během kojení. Cilostazol je zařazen v kategorii C, tj. podezření na teratogenitu vyslovenou na základě preklinických prací při absenci klinických studií v graviditě. Obdobně je kontraindikován při alergii na účinnou látku či na látky pomocné. V pediatrické populaci nebyl efekt cilostazolu prověřován.Dle schválených informací o přípravku (SPC) cilostazol rovněž nepodáváme u renálního či hepatálního selhání. Při sledování hladin cilostazolu a jeho metabolitů u nemocných s renální nedostatečností a s renálním selháním bylo doloženo snížení expozice (AUC) mateřské látce i aktivním dehydrometabolitům o 30–40 % proti zdravým osobám, zvýšení hladiny (na dvojnásobek) bylo zaznamenáno pouze u málo aktivních metabolitů [19]. U hepatálního selhání nebyla farmakokinetika cilostazolu sledována. Důvodem pro kontraindikaci tedy je spíše riziko krvácení nežli zvýšení expozice.
Cilostazol je kontraindikován při současné léčbě silnými inhibitory CYP3A (některá azolová antimykotika, antiretrovirotika, makrolidová antibiotika, zejména klarithromycin) a CYP2C19 (inhibitory protonové pumpy, zejména omeprazol). Při aplikaci středně silných inhibitorů CYP3A4 je doporučeno dávku cilostazolu redukovat na polovinu, tj. 2krát denně 50 mg. Klinicky významnější bude současná aplikace omeprazolu či lansoprazolu, zvýší se totiž nejen expozice samotnému cilostazolu, ale zejména jeho aktivním metabolitům.
Dávkování
Standardní doporučená dávka cilostazolu je 2krát denně 100 mg. Není nutná redukce dávky u starších nemocných ani u jiných subpopulací. Při současné léčbě středně silnými inhibitory oxidázy CYP3A4 (např. amiodaronem, verapamilem, fluoxetinem či grapefruitovou šťávou) je doporučeno snížit dávku na polovinu.
Lékové interakce
Na úrovni interference s farmakodynamickým účinkem je třeba počítat s mírným zvýšením vazodilatačního efektu při kombinaci cilostazolu s blokátory kalciového kanálu či s dalšími vazodilatancii, klinický dopad však bude málo významný. Na rozdíl od inhibitorů PDE-5 není zmiňována interakce cilostazolu s nitráty. Výraznější bude potenciace účinku s antitrombotiky. Jako nejvýznamnější jsou uváděny interakce s antikoagulancii, naopak s protidestičkovými léky je riziko jen středně významné [33].
Farmakokinetické interakce s inhibitory izoenzymu CYP3A4 byly zmíněny. Silné inhibitory, jakými jsou některá azolová antimykotika (ketokonazol, itrakonazol či flukonazol), zvyšují maximální koncentraci i expozici asi dvojnásobně, obdobné zvýšení se předpokládá i u dalších silných inhibitorů (sertralin, fluvoxamin, fluoxetin). Středně silné inhibitory (např. erythromycin, klarithromycin, flavonoidy z citrusů, zejména z grapefruitu, verapamil, diltiazem, amiodaron aj.) zvyšují maximální koncentraci či expozici asi o 50 %. Induktory CYP3A4 (některá antikonvulziva, třezalka aj.) sice snižují expozici mateřské látce, ale na straně druhé zvyšují konverzi na aktivní dehydrometabolity. Výsledný efekt lze těžko predikovat. Inhibitory oxidázy CYP2C19 (zejména omeprazol) zvyšují jak expozici mateřské látce, tak aktivním metabolitům. Cilostazol je slabým inhibitorem CYP3A4, větší klinický význam tato vlastnost nemá.
Závěr
V molekule cilostazolu se nám dostává do rukou účinné léčivo k symptomatické léčbě ICHDK ve stadiu klaudikací. Je pravděpodobné, že díky pozoruhodnému pleiotropnímu účinku nezůstane pouze u této indikace. Probíhají další zajímavé studie v rámci sekundární prevence aterotrombotických příhod.
Práce byla podpořena výzkumným programem Univerzity Karlovy P35.
Seznam použité literatury
- [1] Inoue T, Sohma R, Morooka S. Cilostazol inhibits the expression of activation-dependent membrane surface glycoprotein on the surface of platelets stimulated in vitro. Thromb Res 1999; 93: 137–143.
- [2] Wang S, Cone J, Fong M, et al. Interplay between inhibition of adenosine uptake and phosphodiesterase type 3 on cardiac function by cilostazol, an agent to treat intermittent claudication. J Cardiovasc Pharmacol 2001; 38: 775–783.
- [3] Liu Y, Shakur Y, Kambayashi J. Phosphodiesterases as targets for intermittent claudication. Handb Exp Pharmacol 2011; 204: 211–236.
- [4] Chen H, Wei A, He J, et al. Changes of hypoxia-inducible factor-1 signaling and the effect of cilostazol in chronic cerebral ischemia. Neural Regen Res 2013; 8: 1803–1813.
- [5] Biscetti F, Pecorini G, Straface G, et al. Cilostazol promotes angiogenesis after peripheral ischemia through a VEGF-dependent mechanism. Int J Cardiol 2013; 167: 910–916.
- [6] Phosphodiesterases as Drug Targets, Handbook of Experimental Pharmacology, Volume 204, 2011, Editors: Francis SH, Conti M, Houslay MD, Springer 2011.
- [7] Liu Y, Shakur Y, Yoshitake M, Kambayashi Ji J. Cilostazol (pletal): a dual inhibitor of cyclic nucleotide phosphodiesterase type 3 and adenosine uptake. Cardiovasc Drug Rev 2001; 19: 369–386.
- [8] Lee TM, Lin SZ, Chang NC. Differential effect of phosphodiesterase-3 inhibitors on sympathetic hyperinnervation in healed rat infarcts. Circ J 2014; 78: 366–376.
- [9] Yoshikawa T, Mitani K, Kotosai K, et al. Antiatherogenic effects of cilostazol and probucol alone, and in combination in low density lipoprotein receptor-deficient mice fed with a high fat diet. Horm Metab Res 2008; 40: 473–478.
- [10] Geng DF, Deng J, Jin DM, et al. Effect of cilostazol on the progression of carotid intima-media thickness: a meta-analysis of randomized controlled trials. Atherosclerosis 2012; 220: 177–183.
- [11] Ito H, Uehara K, Matsumoto Y, et al. Cilostazol Inhibits Accumulation of Triglyceride in Aorta and Platelet Aggregation in Cholesterol-Fed Rabbits. PLoS ONE 2012; 7: e39374.
- [12] Ikewaki K, Mochizuki K, Iwasaki M, et al. Cilostazol, a potent phosphodiesterase type III inhibitor, selectively increases antiatherogenic high-density lipoprotein subclass LpA-I and improves postprandial lipemia in patients with type 2 diabetes mellitus. Metabolism 2002; 51: 1348–1354.
- [13] Bramer SL, Forbes WP. Relative bioavailability and effects of a high fat meal on single dose cilostazol pharmacokinetics. Clin Pharmacokinet 1999; 37 (Suppl 2): 13–23.
- [14] Yoo HD, Park SA, Cho HY, et al. Influence of CYP3A and CYP2C19 genetic polymorphisms on the pharmacokinetics of cilostazol in healthy subjects. Clin Pharmacol Ther 2009; 86: 281–284.
- [15] Hiratsuka M, Hinai Y, Sasaki T. Characterization of human cytochrome p450 enzymes involved in the metabolism of cilostazol. Drug Metab Dispos 2007; 35: 1730–1732.
- [16] Suri A, Bramer SL. Effect of omeprazole on the metabolism of cilostazol. Clin Pharmacokinet 1999; 37 (Suppl 2): 53–59.
- [17] Suri A, Forbes WP, Bramer SL. Pharmacokinetics of multiple-dose oral cilostazol in middle-age and elderly men and women. J Clin Pharmacol 1998; 38: 144–150.
- [18] Schrör K. The pharmacology of cilostazol. Diabetes Obes Metab 2002; 4 (Suppl 2): S14–19.
- [19] Mallikaarjun S, Forbes WP, Bramer SL, et al. Effect of renal impairment on the pharmacokinetics of cilostazol and its metabolites. Clin Pharmacokinet 1999; 37 (Suppl 2): 33–40.
- [20] Thompson PD, Zimet R, Forbes WP, et al. Metaanalysis of results from eight randomized, placebo-controlled trials on the effect of cilostazol on patients with intermittent claudication. Am J Cardiol 2002; 90: 1314–1319.
- [21] Mohler ER, Beebe HG, Salles-Cuhna S, et al. Effects of cilostazol on resting ankle pressures and exercise-induced ischemia in patients with intermittent claudication. Vasc Med 2001; 6: 151–156.
- [22] Warner CJ, Greaves SW, Larson RJ, et al. Cilostazol is associated with improved outcomes after peripheral endovascular interventions. J Vasc Surg 2014; 59: 1607–1614.
- [23] Dawson DL, Cutler BS, Hiatt WR. A comparison of cilostazol and pentoxifylline for treating intermittent claudication. Am J Med 2000; 109: 523–530.
- [24] Bedenis R, Stewart M, Cleanthis M, et al. Cilostazol for intermittent claudication (Review). The Cochrane Library 2014; Issue 10. DOI: 10.1002/14651858.CD003748.pub4
- [25] Stevens JW, Simpson E, Harnan S, et al. Systematic review of the efficacy of cilostazol, naftidrofuryl oxalate and pentoxifylline for the treatment of intermittent claudication. Br J Surg 2012; 99: 1630–1638.
- [26] Matsumoto M. Cilostazol in secondary prevention of stroke: impact of the Cilostazol Stroke Prevention Study. Atheroscler Suppl 2005; 6: 33–40.
- [27] Shinohara Y, Katayama Y, Uchiyama S, et al. Cilostazol for prevention of secondary stroke (CSPS 2): an aspirin-controlled, double-blind, randomised non-inferiority trial. Lancet Neurol 2010; 9: 959–968.
- [28] Huang Y, Cheng Y, Wu J, et al. Cilostazol as an alternative to aspirin after ischaemic stroke: a randomised, double-blind, pilot study. Lancet Neurol 2008; 7: 494–499.
- [29] Hiatt WR, Money SR, Brass EP. Long-term safety of cilostazol in patients with peripheral artery disease: the CASTLE study (Cilostazol: A Study in Long-term Effects). J Vasc Surg 2008; 47: 330–336.
- [30] Woo SK, Kang WK, et al. Pharmacokinetic and pharmacodynamic modeling of the antiplatelet and cardiovascular effects of cilostazol in healthy humans. Clin Pharmacol Ther 2002; 71: 246–252.
- [31] Christofi FL. Purinergic receptors and gastrointestinal secretomotor function. Purinergic Signal 2008; 4: 213–236.
- [32] http://www.drugs.com/drug-interactions/cilostazol,pletal.html (Navštíveno 20. 11. 2014.)