Durvalumab
Souhrn:
Bílek O. Durvalumab. Remedia 2020; 30: 371–374.
Durvalumab je plně humanizovaná IgGκ monoklonální protilátka, která selektivně blokuje ligand receptoru programované buněčné smrti (PD‑L1). V Evropské unii je registrován ke konsolidační léčbě lokálně pokročilého inoperabilního nemalobuněčného karcinomu plic (NSCLC) bez progrese po chemoradiační terapii (CRT).
Summary:
Bilek O. Durvalumab. Remedia 2020; 30: 371–374.
Durvalumab is a fully humanized IgGκ monoclonal antibody that selectively blocks programmed cell death ligand 1 (PD‑L1). In European Union, it is registered for consolidation treatment of non‑small cell lung cancer (NSCLC) which has not progressed following platinum‑based chemoradiation therapy (CRT).
Key words: durvalumab, non‑small cell lung cancer, immunotherapy
Úvod
Durvalumab (Imfinzi; MEDI4736) je plně humanizovaná IgGκ monoklonální protilátka, která selektivně blokuje ligand receptoru programované buněčné smrti 1 (programmed cell death ligand 1, PD L1). Vazbou na PD L1 blokuje interakci s receptorem programované buněčné smrti 1 (programmed cell death protein 1, PD 1) a CD80 a tím umožní obnovu imunitní protinádorové reakce a terapeutický efekt [1]. V Evropské unii je durvalumab registrován k léčbě lokálně pokročilého neoperovatelného nemalobuněčného karcinomu plic (non small cell lung cancer, NSCLC) s expresí PD L1 alespoň 1 % v rámci konsolidační imunoterapie po chemoradioterapii na bázi platiny. V České republice není k 30. 7. 2020 léčba durvalumabem hrazena.
Mechanismus účinku
Proteinový receptor PD 1 je
exprimován na aktivovaných cytotoxických T lymfocytech. Jsou
známy dva ligandy – PD L1 (B7 H1, CD274) a PD L2
(B7 DC, CD273), které jsou exprimovány na T lymfocytech,
B lymfocytech, antigen prezentujících (APC) a stromálních
buňkách a mohou být exprimovány rovněž na nádorových
buňkách. PD L1 má vazbu buď k PD 1, nebo k CD80
(B7 1) exprimovanému na aktivovaných T lymfocytech nebo
APC. Interakce mezi PD L1 a PD 1/CD80 vede k inhibici
aktivace T lymfocytů a k přerušení imunitní odpovědi
[2]. Nádorové buňky, které exprimují PD L1, mohou
prostřednictvím tohoto receptoru vypnout imunitní reakci
cytotoxických T lymfocytů vůči nim a znemožnit tak imunitní
dohled nad nádorovými buňkami. Durvalumab selektivně blokuje
interakci mezi PD L1 a PD 1/CD80 a zabrání
utlumení imunitní reakce (obr. 1).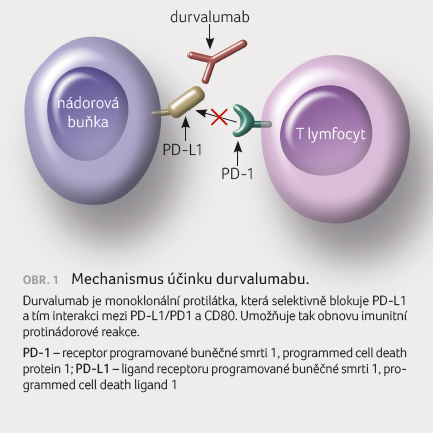
Farmakokinetika
Durvalumab byl v rámci klinických studií dávkován v rozmezí 0,1‒20 mg/kg, následně byla stanovena terapeutická dávka 10 mg/kg. Měl by být podáván v samostatné intravenózní infuzi trvající 60 minut. Jeho biotransformace nesouvisí s enzymy, které běžně metabolizují léčiva včetně cytochromu P450. Nevyvolává inhibici ani indukci těchto enzymů. Primárními eliminačními cestami durvalumabu jsou katabolismus proteinů v retikuloendotelovém systému nebo přeměny v cílových buňkách. Nejsou proto očekávány metabolické interakce s jinými léčivy. Nebyly provedeny žádné studie, které by zkoumaly inkompatibilitu s jinými látkami. Je možné ovlivnění farmakodynamiky durvalumabu u pacientů, jimž byly podány systémové kortikosteroidy (≥ 10 mg/den prednisonu nebo ekvivalentní) či jiná imunosupresiva před zahájením léčby. Je zde možnost interference s mechanismem účinku durvalumabu, nikoliv však s jeho farmakokinetikou.
Durvalumab v léčbě lokálně
pokročilého NSCLC
Pacienti s lokálně pokročilým inoperabilním NSCLC jsou standardně léčeni radioterapií a chemoterapií založenou na derivátu platiny. Konkomitantní aplikace chemoterapie/radioterapie (chemoradiotherapy, CRT) byla spojena s lepšími výsledky léčby [3].
Preklinická data konzistentně ukazují prospěch z kombinace anti PD L1 imunoterapie s radioterapií [4]. Radioterapie se podílí na redukci nádorových buněk prostřednictvím apoptózy, nekrózy, mitotické katastrofy, autofagie či stárnutí (senescence). Prostřednictvím těchto mechanismů může „zviditelnit“ nádorové antigeny pro vrozené a adaptivní složky imunitního systému. Dochází k aktivaci cytotoxických T lymfocytů či ke zvýšené expresi hlavního histokompatibilního komplexu (major histocompatibility complex, MHC) I. třídy [5]. Indukcí reakce T lymfocytů proti specifickému antigenu může navodit léčebnou odpověď i mimo ozařované pole známou jako abskopální efekt [6]. Poškození nádorové DNA bývá spojeno se vznikem neoantigenů a následná imunitní reakce může vést k obnovení imunitního dohledu.
Multicentrická dvojitě zaslepená studie fáze III PACIFIC (NCT02125461) hodnotila efekt konsolidační léčby durvalumabem po standardní konkomitantní CRT u pacientů s lokálně pokročilým inoperabilním NSCLC třetího klinického stadia [7].
Zařazení pacienti absolvovali dva či více cyklů chemoterapie založené na platinovém derivátu konkomitantně s definitivní radioterapií v dávce 54‒66 Gy. Bylo zařazeno 713 pacientů, kteří byli randomizováni v poměru 2 : 1 ve prospěch imunoterapie. Pokud nedošlo k progresi onemocnění, byla v horizontu 1‒42 dní po ukončení CRT zahájena konsolidační aplikace durvalumabu v dávce 10 mg/kg tělesné hmotnosti ve dvoutýdenním intervalu, nebo placeba. Primární cílové ukazatele představovaly celkové přežití (overall survival, OS) a přežití bez progrese (progression free survival, PFS). Sekundárními cílovými ukazateli byly PFS ve 12 a 18 měsících, četnost objektivních odpovědí (objective response rate, ORR), doba trvání odpovědi, doba do úmrtí/rozvoje vzdálených metastáz, OS ve 24 měsících, čas do druhé progrese onemocnění, bezpečnost léčby, kvalita života a farmakokinetika. Součástí studie bylo i stanovení exprese PD L1 v nádorové tkáni, stav exprese však neměl vliv na zařazení pacientů.
První interim analýza prokázala delší medián PFS 16,8 měsíce vs. 5,6 měsíce ve prospěch durvalumabu (poměr rizik [HR] 0,52; 95% interval spolehlivosti [CI] 0,42–0,65; p < 0,001). Dále byla doložena vyšší ORR ve skupině s durvalumabem oproti skupině s placebem – 28,4 % vs. 16 % (p < 0,001). V této analýze byl významně prodloužen čas do druhé progrese nebo do úmrtí ve skupině s durvalumabem (medián 28,3 měsíce vs. 17,1 měsíce) [8]. Durvalumab však zejména přinesl významné zlepšení OS; v rámci tříletého sledování nebylo dosaženo mediánu OS v rameni s durvalumabem vs. 29,1 měsíce v rameni s placebem. Ve 12, 24 a 36 měsících přežívalo 83,1 % vs. 74,6 %, 66,3 % vs. 55,3 % a 57,0 % vs. 43,5 % pacientů ve prospěch durvalumabu (HR 0,69; 95% CI 0,55‒0,86) [9]. Prodloužení PFS a OS bylo zaznamenáno bez ohledu na použitý režim chemoterapie, dávku radioterapie nebo dobu od radioterapie do randomizace. Zlepšení sledovaných parametrů bylo zaznamenáno u všech podskupin včetně pacientů s nízkou expresí PD L1 [10]. V roce 2018 však byla prezentována post hoc analýza s cílem posouzení vlivu exprese PD L1 na medián PFS. Ve skupině pacientů s PD L1 alespoň 1 % bylo dosaženo mediánu PFS (median progression free survival, mPFS) 17,8 měsíce v rameni s léčbou durvalumabem oproti 5,6 měsíce při podávání placeba. U pacientů s expresí PD L1 méně než 1 % bylo v rameni s durvalumabem dosaženo mPFS 10,7 měsíce, zatímco v rameni s placebem 5,6 měsíce (HR 0,73; 95% CI 0,48–1,1). U 37 % pacientů nebyla míra exprese známa. V rámci Evropské unie byla schválena konsolidační léčba durvalumabem pro pacienty s expresí PD L1 alespoň 1 % [11].
Bezpečnost a snášenlivost
Bezpečnost durvalumabu v dávce 10 mg/kg byla hodnocena ve studii PACIFIC v rámci konsolidační léčby u pacientů s lokálně pokročilým inoperabilním karcinomem plic po CRT. Nejčastějším nežádoucím účinkem stupně 3‒4 byla pneumonie. Podávání durvalumabu je nejčastěji spojeno s imunitně podmíněnými nežádoucími účinky: pneumonitida nebo intersticiální plicní nemoc, hepatitida, kolitida, hypotyreóza, hypertyreóza, insuficience nadledvin, diabetes mellitus 1. typu, hypofyzitida/hypopituitarismus, nefritida, vyrážka. Imunitně podmíněné nežádoucí účinky 2.‒4. stupně vedou k přerušení léčby durvalumabem, výjimkou jsou endokrinopatie [12]. Většina imunitně podmíněných nežádoucích účinků, včetně závažných, odezněla po zahájení vhodné léčby nebo po přerušení podávání přípravku. Léčba spočívá zpravidla v aplikaci kortikosteroidů, ev. při nezmírnění symptomů terapie pokračuje podáváním dalších imunosupresiv jako infliximabu nebo mykofenolát mofetilu [13]. Po zlepšení stavu je nutné snižovat/vysazovat dávku kortikosteroidů po dobu 4‒6 týdnů.
Imunitně podmíněná pneumonitida nebo intersticiální plicní onemocnění je závažným nežádoucím účinkem, který může ohrozit život pacienta. Klinicky a podle zobrazovacích metod se podobá radiační pneumonitidě pozorované u pacientů podstupujících radioterapii plic. Klinickými projevy jsou dušnost a hypoxie. Je nutné vyloučení infekční příčiny potíží včetně zvážení bronchoskopie s vyšetřením bronchoalveolární laváže. Je vhodná empirická antibiotická léčba. V případě pneumonitidy 2. stupně je indikováno přerušení léčby durvalumabem a zahájení léčby kortikosteroidy v dávce odpovídající 1‒2 mg/kg prednisonu denně. Po zlepšení nálezu a postupné redukci dávky kortikosteroidů lze zvážit pokračování léčby durvalumabem. Pneumonitida stupně 3 nebo 4 vyžaduje trvalé přerušení léčby a zahájení kortikoterapie v dávce odpovídající 1‒4 mg/kg prednisonu za den. Pokud po 48 hodinách nedojde ke zlepšení, lze volit mezi terapií dalšími imunosupresivy jako infliximabem nebo intravenózním mykofenolát mofetilem.
Imunitně podmíněná hepatitida se projevuje zvýšením hodnot aminotransferáz, popř. celkového bilirubinu bez souvislosti s infekční hepatitidou nebo základním onkologickým onemocněním. V případě hepatitidy 2. stupně je indikováno přerušení léčby durvalumabem, pokud nedojde ke zlepšení hodnot jaterních testů, je nutné zahájení léčby kortikosteroidy v dávce odpovídající 1‒2 mg prednisonu za den. Po úpravě hodnot a postupné redukci dávky kortikosteroidů lze zvážit pokračování léčby durvalumabem. Hepatitida 3. a 4. stupně vyžaduje trvalé ukončení léčby durvalumabem a zahájení kortikoterapie odpovídající podání 1‒2 mg/kg prednisonu za den.
Imunitně podmíněná kolitida se projevuje závažnými průjmy a dalšími symptomy, jako jsou bolesti břicha, příměs hlenu či krve ve stolici. Musí být vyloučena infekční příčina potíží. U průjmu nebo kolitidy 2. stupně je indikováno přerušení léčby durvalumabem a při přetrvávání potíží zahájení kortikoterapie v dávce odpovídající 1‒2 mg/kg prednisonu za den. Po odeznění potíží a postupné redukci dávky kortikosteroidů lze zvážit pokračování léčby durvalumabem. V případě kolitidy či průjmu 3. a 4. stupně je indikováno trvalé přerušení léčby durvalumabem a zahájení kortikoterapie odpovídající podání 1‒2 mg/kg prednisonu za den, popř. dalšími imunosupresivy při nezlepšení stavu.
Imunitně podmíněná nefritida se projevuje asymptomatickým zvýšením hodnoty kreatininu v séru, které nesouvisí se základním onemocněním. Při zvýšení hodnoty na 2. stupeň je indikováno přerušení léčby durvalumabem, nedojde li k úpravě, je indikována léčba kortikosteroidy v dávce odpovídající 1‒2 mg/kg prednisonu za den. Při nefritidě 3. a 4. stupně je indikováno trvalé přerušení léčby durvalumabem a zahájení léčby kortikosteroidy odpovídající aplikaci 1‒2 mg/kg prednisonu denně.
Imunitně podmíněná endokrinopatie se projevuje nejčastěji jako hypotyreóza, hypertyreóza, hypofyzitida, funkční nedostatečnost nadledvin a diabetická ketoacidóza. Pacienti léčení durvalumabem musejí být před léčbou a v jejím průběhu monitorováni pomocí biochemických parametrů.
Klinicky se toxicita projevuje únavou, bolestmi hlavy, změnami duševního stavu, bolestmi břicha, neobvyklými střevními projevy a hypotenzí nebo nespecifickými symptomy, které se mohou podobat přítomnosti mozkových metastáz. Je nutné vyloučit souvislost se základním onemocněním. Přerušení léčby durvalumabem se doporučuje jen u symptomatické endokrinopatie do stabilizace klinického stavu. Vzhledem k vysokému riziku vzniku imunitně podmíněné tyreopatie patří k základnímu vyšetření před zahájením a v průběhu léčby monitorace hodnot tyreotropního hormonu (TSH) a volného tyroxinu (fT4). V případě hypotyreózy je nutné zahájit substituční léčbu hormony štítné žlázy, u hypertyreózy lze zvážit léčbu methimazolem. Hypertyreóza často během několika týdnů přechází do hypotyreózy. Při symptomatické nedostatečnosti nadledvin je indikováno přerušení léčby durvalumabem a nastavení substituce kortikosteroidů. V případě symptomatické hypofyzitidy se po přerušení podávání durvalumabu musí zahájit substituční hormonální léčba. Při podezření na akutní zánět hypofýzy je nutno zvážit podání kortikosteroidů v dávce odpovídající 1‒2 mg/kg prednisonu denně. Pokud dojde ke zlepšení stavu, lze po postupném utlumení užívání kortikosteroidů pokračovat v léčbě durvalumabem. Nadále je nutné monitorovat funkce hypofýzy a sledovat hormonální parametry k zajištění odpovídající hormonální substituční léčby. V případě rozvoje diabetu je nutné zahájit substituční léčbu inzulinem za monitorace glykemie.
Imunitně podmíněná kožní toxicita se projevuje různými typy vyrážky. Podávání durvalumabu má být přerušeno při vyrážce 2. stupně trvající déle než jeden týden a při vyrážce 3. stupně, ukončeno pak při 4. stupni. Závažnou vyrážku léčíme vysokými dávkami kortikosteroidů v dávce odpovídající 1‒2 mg/kg prednisonu denně.
V průběhu léčby durvalumabem se mohou vyskytnout další vzácné imunitně podmíněné nežádoucí účinky. U méně než 1 % pacientů byly zaznamenány myasthenia gravis, myokarditida, myozitida, polymyozitida.
Při výskytu nežádoucích účinků v průběhu aplikace durvalumabu je nutné terapii přerušit. Pokud se jedná o reakci nižšího stupně, lze v léčbě pokračovat za přísné monitorace, v případě závažné reakce 4. stupně musí být terapie ukončena.
Kontraindikace
Kontraindikací aplikace durvalumabu je hypersenzitivita na léčivou látku nebo na kteroukoliv látku uvedenou v souhrnu údajů o přípravku [14].
Popis přípravku
Durvalumab je dodáván jako čirý až opalizující, bezbarvý až světle žlutý koncentrát pro infuzní roztok. Jeden ml koncentrátu obsahuje 50 mg durvalumabu. Roztok má pH přibližně 6,0 a osmolalitu přibližně 400 mosm/kg. Je dostupný v baleních 120 mg a 500 mg. Doporučená dávka přípravku je 10 mg/kg aplikovaná v samostatné intravenózní infuzi trvající 60 minut jedenkrát za dva týdny až do progrese onemocnění nebo do nepřijatelné toxicity nebo maximálně po dobu 12 měsíců. Zvyšovat nebo snižovat dávku se nedoporučuje. Ukončení nebo přerušení léčby je závislé na individuální bezpečnosti a snášenlivosti.
Závěr
Konsolidační léčba durvalumabem po definitivní CRT u pacientů s inoperabilním NSCLC třetího klinického stadia s PD L1 alespoň 1 % se stává součástí standardního léčebného algoritmu. Durvalumab je referenční molekulou v této indikaci. Léčba je spojena s dobrou tolerancí a příznivým bezpečnostním profilem.
Seznam použité literatury
- [1] Stewart R, Morrow M, Hammond SA, et al. Identification and Characterization of MEDI4736, an Antagonistic Anti‑PD‑L1 Monoclonal Antibody. Cancer Immunol Res 2015; 3: 1052–1062.
- [2] Lee HT, Lee JY, Lim H, et al. Molecular Mechanism of PD‑1/PD‑L1 Blockade via Anti‑PD‑L1 Antibodies Atezolizumab and Durvalumab. Sci Rep 2017; 7: 5532.
- [3] Aupérin A, Le Péchoux C, Rolland E, et al. Meta‑Analysis of Concomitant versus Sequential Radiochemotherapy in Locally Advanced Non‑Small‑Cell Lung Cancer. J Clin Oncol 2010; 28: 2181–2190.
- [4] Formenti SC, Demaria S. Combining Radiotherapy and Cancer Immunotherapy: A Paradigm Shift. J Natl Cancer Inst 2013; 105: 256–265.
- [5] Golden EB, Pellicciotta I, Demaria S, et al. The Convergence of Radiation and Immunogenic Cell Death Signaling Pathways. Front Oncol 2012; 2: 88.
- [6] Demaria S, Ng B, Devitt ML, et al. Ionizing Radiation Inhibition of Distant Untreated Tumors (Abscopal Effect) Is Immune Mediated. Int J Radiat Oncol Biol Phys 2004; 58: 862–870.
- [7] Planchard D, Popat S, Kerr K, et al. Metastatic Non‑Small Cell Lung Cancer: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow‑Up. Ann Oncol 2019; 30: 863–870.
- [8] Antonia SJ, Villegas A, Daniel D, et al.; PACIFIC Investigators. Durvalumab after Chemoradiotherapy in Stage III Non‑Small‑Cell Lung Cancer. N Engl J Med 2017; 377: 1919–1929.
- [9] Gray JE, Villegas A, Daniel D, et al. Three‑Year Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC‑Update from PACIFIC. J Thorac Oncol 2020; 15: 288‒293.
- [10] Faivre‑Finn C, Spigel DR, Senan S, et al. Efficacy and Safety Evaluation Based on Time from Completion of Radiotherapy to Randomization with Durvalumab or Placebo in Pts from PACIFIC. Ann Oncol 2018; 29(Suppl 8): viii488‒viii492.
- [11] European Medical Agency. Imfinzi: Summary of Product Characteristics. Dostupné na: https://www.ema.europa.eu/en/documents/product‑inf
- [12] NCCN Guidelines. Management of Immunotherapy‑Related Toxicities. Version 1.2020 – December 16, 2019. Dostupné na: https://www.nccn.org
- [13] Darnell EP, Mooradian MJ, Baruch EN, et al. Immune‑Related Adverse Events (IrAEs): Diagnosis, Management, and Clinical Pearls. Curr Oncol Rep 2020; 22: 39.
- [14] IMFINZI (durvalumab). SPC 2020. Dostupné na: https://www.ema.europa.eu/en/documents/product‑information/imfinzi‑epar‑product‑information_en.pdf