Siponimod
Souhrn:
Kubala Havrdová E. Siponimod. Remedia 2020; 30: 297–304.
Roztroušená skleróza (RS) je zánětlivé chronické onemocnění centrálního nervového systému s autoimunitními pochody v patogenezi vedoucí bez léčby k závažné invaliditě mladých nemocných. Siponimod je selektivní modulátor sfingosin‑1‑fosfátových (S1P) receptorů, který byl úspěšně testován jak u relabující‑remitující RS, tak u sekundární progrese. Jde o první perorální lék schválený pro použití u sekundární progrese, zřejmě pro jeho schopnost nejen procházet hematoencefalickou bariérou, ale i pro jeho vazbu na S1P receptory v mozku a schopnost působit tak na neurodegenerativní procesy spojené s progresí invalidity u RS, nejen pouze na děje zánětlivé. Ty jsou nejvíce aktivní v začátku nemoci a v současné době máme již celou paletu protizánětlivých léků schválených pro léčbu relabující‑remitující fáze nemoci. Bezpečnost siponimodu v základní registrované dávce 2 mg p.o. denně se v zásadě neliší od dosud schválených přípravků této skupiny, modulátorů S1P receptorů. Výhodou je selektivita pro receptory S1P1 a S1P5 a omezení kardiálních nežádoucích účinků. Titrace dávky je uživatelsky příjemná pro pacienta i lékaře, a u většiny pacientů tak odpadá nutnost monitorace zahajovací dávky pro možnou bradykardii.
Summary:
Kubala Havrdova E. Siponimod. Remedia 2020; 30: 297–304.
Multiple sclerosis is a chronic inflammatory disease of the central nervous system with autoimmune processes in pathogenesis, leading to substantial disability in young adults if untreated. Siponimod is a selective modulator of sfingosin‑1‑phosphate (S1P) receptors that was successfully investigated in both relapsing‑remitting and secondary progressive multiple sclerosis. It is the first approved oral medication in secondary progression, obviously for its ability not only to cross the blood‑brain barrier but also to bind the S1P receptors in the brain, and for its ability to influence neurodegenerative processes which are responsible for disability progression in multiple sclerosis. Therefore, siponimod acts not only on inflammatory processes that are more connected with the onset of the disease. Currently, the inflammation in relapsing‑remitting multiple sclerosis can be treated with a number of approved drugs. The safety of the basic registered dose of siponimod of 2 mgs resembles the safety of other registered drugs in this class (S1P receptor modulators). The advantage is the decrease of cardiac side effects and selectivity for S1P1 and S1P5 receptors. Titration of the dose is user friendly both for patients and physicians. Monitoring the dose for possible bradycardia is not necessary anymore.
Key words: siponimod, multiple sclerosis, sfingosin‑1‑phospate receptors, secondary progression
Siponimod (Mayzent) je řazen
do farmakologické skupiny Imunosupresiva, selektivní
imunosupresiva, ATC kód: L04AA42. Jeho indikací je sekundárně
progresivní roztroušená skleróza (SP RS) s aktivním
onemocněním doloženým relapsy nebo zánětlivou aktivitou pomocí
zobrazovacích metod.
Dávkování
Standardní dávkování představuje 2 mg siponimodu p.o. denně. Před zahájením léčby je však nutno provést genotypizaci pro enzym cytochrom P450 2C9 (CYP2C9) z důvodu zjištění typu metabolizéra. Pacienti s genotypem CYP2C9*3*3 by neměli siponimod užívat vůbec, pacienti s genotypem CYP2C9*2*3 nebo *1*3 jsou léčeni poloviční dávkou 1 mg denně.
Léčba se zahajuje titračním balením pro prvních pět dnů: první a druhý den 0,25 mg, třetí den 0,5 mg, čtvrtý den 0,75 mg, pátý den 1,25 mg. Od šestého dne užívá pacient udržovací dávku 2 mg nebo 1 mg. Je li léčba přerušena na čtyři a více dnů, je nutno provést znovu titraci [1].
Zařazení
do současné palety léčiv
Roztroušená skleróza je imunitně zprostředkované onemocnění centrálního nervového systému (CNS). Zánětlivá demyelinizační ložiska v CNS vedou k různým neurologickým příznakům. Začátek nemoci je typicky charakterizován atakami neurologické symptomatologie s následným zlepšením, během života však dochází k akumulaci demyelinizace a axonální ztráty a nárůstu invalidity. V tomto progresivním stadiu se patogeneticky uplatňují neurodegenerativní procesy i pokračující chronický zánět, převážně již kompartmentalizovaný za hematoencefalickou bariérou [2].
Pro časná stadia onemocnění existuje již řada léků (interferon beta, glatiramer acetát, teriflunomid, dimetylfumarát, fingolimod, kladribin, natalizumab, alemtuzumab, okrelizumab) zaměřených především proti zánětu, které omezují počet relapsů a vznik nových lézí v CNS, a ty účinnější oddalují i nástup klinické progrese nemoci. V relapsech dochází k ničení myelinu i axonů, čím účinněji je zánět potlačen, tím pomaleji onemocnění postupuje.
Patogeneze progrese není plně prozkoumána, nacházíme zde jednak mechanismy vrozené imunity, uplatňují se ale i faktory jako oxidační stres, poškození mitochondrií, poruchy iontových kanálů na poškozených axonech, akumulace železa v extracelulárním prostoru podílející se na destrukci tkáně pomocí volných radikálů, difuzní aktivace mikroglie, astrocytární glióza [3]. To vše vede k pozvolnému horšení funkce CNS.
Pro progresivní RS byla zkoušena řada léků, z nichž pouze mitoxantron a okrelizumab byly schváleny regulatorními úřady, přičemž mitoxantron se již téměř nepoužívá kvůli závažné kardiotoxicitě a s léčbou spojené sekundární leukemii. Okrelizumab (protilátka proti CD20) byl testován a schválen u populace primárně progresivní RS (PP RS). Je schopen zpomalit progresi. Jinak je hojně používán u relabující-remitující RS (RR-RS) jako lék pro nemoc s vyšší aktivitou.
Natalizumab, velmi účinný lék u RR-RS, byl testován u SP RS, ale nebyl schválen, prokázal však schopnost zlepšit funkci horní končetiny.
Fingolimod, první schválený modulátor sfingosin-1-fosfátových (S1P) receptorů pro RR-RS, byl také testován u PP RS, nebyl však úspěšný [4].
Siponimod byl ve studii fáze II zkoušen u RR-RS s velmi dobrým efektem srovnatelným s efektem fingolimodu, III. fáze klinického zkoušení proběhla u SP RS, fáze onemocnění, pro niž zatím žádná schválená léčba neexistovala. Siponimod prokázal schopnost zpomalit sekundární progresi RS. Bezpečnostní profil je velmi podobný fingolimodu.
Invalidita pacientů v sekundární progresi RS jim většinou znemožňuje práceschopnost. Udržení soběstačnosti je však z hlediska nákladové efektivity léčby velmi důležité, protože největší náklady u RS nese těžká invalidita pozdních stadií [5].
Mechanismus
účinku, farmakodynamický efekt
Siponimod je modulátorem receptoru pro
S1P se specifickou selektivitou pro dva z pěti receptorů
spřažených s G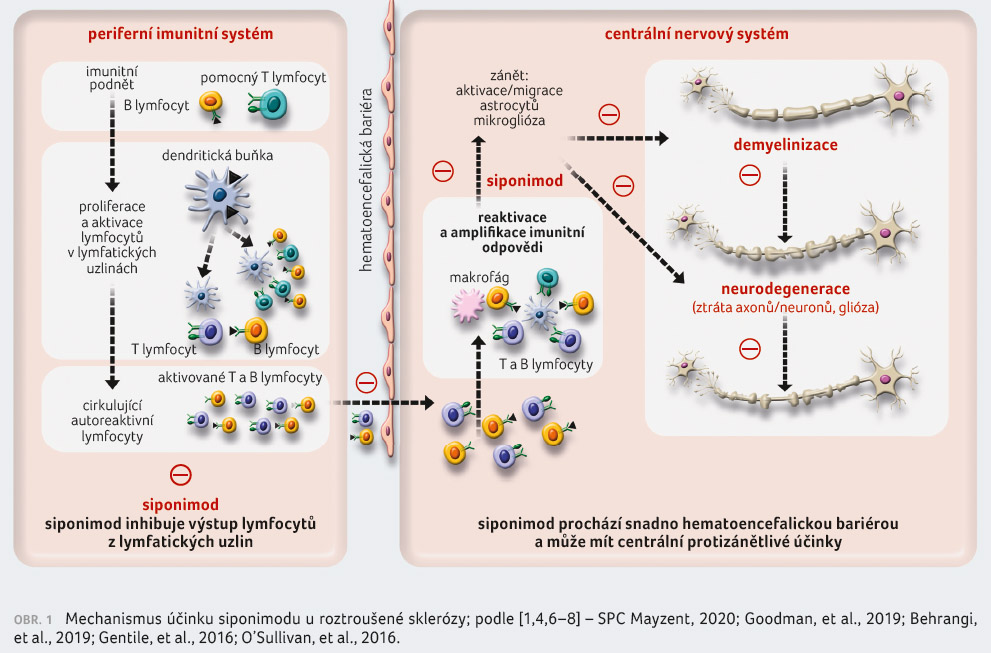 proteinem pro S1P, jmenovitě S1P1
a S1P5 [1]. Tyto receptory jsou
exprimovány na buňkách imunitního systému a CNS [4,6].
Působením jako funkční antagonista S1P1 receptoru
na lymfocytech blokuje jejich schopnost vystupovat
z lymfatických uzlin. Ke snížení počtu lymfocytů
v periferní krvi tak dochází v důsledku reverzibilní
sekvestrace lymfocytů v lymfoidních tkáních. Tím je omezena
recirkulace T lymfocytů do CNS, což nepřímo vede k omezení
zánětu v CNS. Preklinické údaje naznačují, že působením
na receptory S1P1 a S1P5 na buňkách
CNS může siponimod mít i přímé protizánětllivé účinky
v CNS [1,4,6‒8] (obr. 1, 2).
proteinem pro S1P, jmenovitě S1P1
a S1P5 [1]. Tyto receptory jsou
exprimovány na buňkách imunitního systému a CNS [4,6].
Působením jako funkční antagonista S1P1 receptoru
na lymfocytech blokuje jejich schopnost vystupovat
z lymfatických uzlin. Ke snížení počtu lymfocytů
v periferní krvi tak dochází v důsledku reverzibilní
sekvestrace lymfocytů v lymfoidních tkáních. Tím je omezena
recirkulace T lymfocytů do CNS, což nepřímo vede k omezení
zánětu v CNS. Preklinické údaje naznačují, že působením
na receptory S1P1 a S1P5 na buňkách
CNS může siponimod mít i přímé protizánětllivé účinky
v CNS [1,4,6‒8] (obr. 1, 2).
Pokles hodnoty lymfocytů v krvi
je závislý na dávce a nastává během šesti hodin
po první dávce. Při pokračujícím denním dávkování
tento pokles pokračuje a dosahuje nejnižšího mediánu (90%
interval spolehlivosti [CI]) počtu lymfocytů přibližně 0,560
(0,271‒1,08) buněk/nl u typického genotypu CYP2C9*1*1
nebo *1*2 u pacienta se SP RS, odpovídajícího 20‒30 %
hodnot před léčbou [1].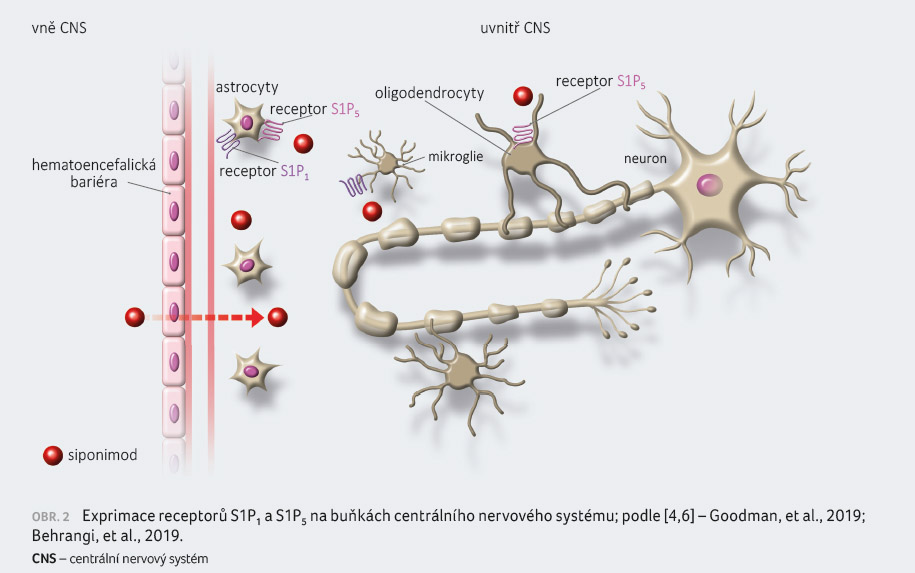
U převážné většiny (90 %) pacientů se SP RS se počet lymfocytů vrací do normálního rozmezí během 10 dnů od ukončení léčby. Reziduální vliv na snížení počtu periferních lymfocytů může přetrvávat po dobu 3‒4 týdnů od poslední dávky [1].
Siponimod se v řadě parametrů
odlišuje od S1P modulátoru fingolimodu, který se používá
v klinické praxi [1,9] (tab. 1).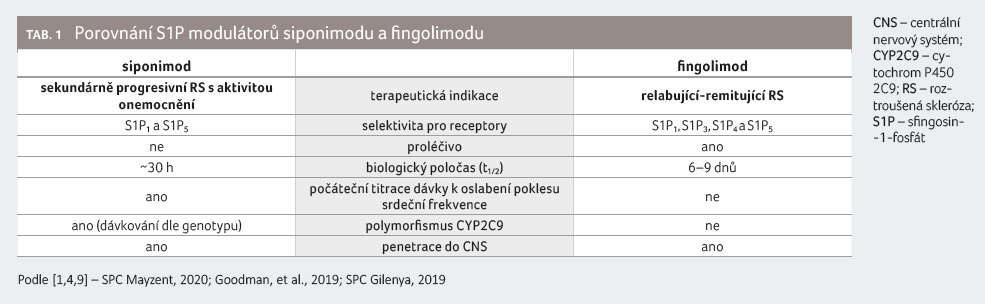
Farmakokinetické
vlastnosti
Absorpce
Doba (tmax) k dosažení maximálních plazmatických koncentrací (cmax) po opakovaném perorálním podání siponimodu je přibližně čtyři hodiny (rozmezí 2‒12 hodin). Absorpce je rozsáhlá a absolutní perorální biologická dostupnost siponimodu činí přibližně 84 %. Při podávání 2 mg siponimodu jedenkrát denně po dobu 10 dnů byla pozorována průměrná cmax 30,4 ng/ml a průměrná plocha pod křivkou plazmatické koncentrace (AUCtau) 558 h.ng/ml v den 10. Rovnovážného stavu bylo dosaženo přibližně po šesti dnech opakovaného podávání siponimodu jednou denně. Přes prodloužení tmax na osm hodin po jednorázovém podání příjem potravy neovlivňuje systémovou expozici siponimodu (cmax a AUC), proto může být siponimod užíván bez ohledu na jídlo [1].
Distribuce
Siponimod je distribuován do tělesných tkání s průměrným distribučním objemem 124 litrů. Jeho frakce v plazmě je u člověka 68 %. Vazba siponimodu na proteiny činí > 99,9 % u zdravých jedinců a u pacientů s poruchou funkce jater nebo ledvin. Siponimod snadno přestupuje hematoencefalickou bariéru [1].
Biotransformace
Siponimod je extenzivně metabolizován, převážně CYP2C9 (79,3 %) a v menší míře CYP3A4 (18,5 %). Neočekává se, že by farmakologická aktivita hlavních metabolitů M3 a M17 měla vliv na klinický účinek a bezpečnost siponimodu u člověka [1].
CYP2C9 je polymorfní a genotyp ovlivňuje dílčí příspěvek dvou oxidačních metabolických cest k celkové eliminaci. Fyziologicky založené farmakokinetické modelování naznačuje rozdílnou, na genotypu CYP2C9 závislou inhibici a indukci CYP3A4. Při snížené metabolické aktivitě CYP2C9 u příslušných genotypů se předpokládá větší účinek substrátů CYP3A4 na expozici siponimodu [1].
Eliminace
U pacientů s RS byla zjištěna zdánlivá systémová clearance (CL/F) 3,11 l/h. Zdánlivý poločas eliminace siponimodu je přibližně 30 hodin. Siponimod je ze systémového oběhu odstraňován hlavně metabolismem a následným vylučováním žlučí/stolicí. Nezměněný siponimod nebyl v moči detekován [1].
Charakteristiky zvláštních skupin pacientů ‒ genotyp CYP2C9
Genotyp CYP2C9 ovlivňuje CL/F siponimodu. Dvě populační farmakokinetické analýzy ukázaly, že subjekty s CYP2C9*1*1 a *1*2 se chovají jako silní metabolizéři, subjekty s *2*2 a *1*3 jako středně silní metabolizéři a subjekty s *2*3 a *3*3 jako slabí metabolizéři. Ve srovnání se subjekty s CYP2C9*1*1 mají jednotlivci s genotypy CYP2C9*2*2, *1*3, *2*3 a *3*3 o 20 %, 35‒38 %, 45‒48 %, resp. 74 % nižší hodnoty CL/F. Expozice siponimodu je proto přibližně o 25 %, 61 %, 91 %, resp. o 284 % vyšší u subjektů s CYP2C9*2*2, *1*3, *2*3 a *3*3, v porovnání se subjekty s *1*1 [1].
Klinické
zkušenosti
Siponimod byl testován u RR-RS ve fázi II (BOLD) [10] a byla hledána nejvhodnější dávka. Studie zahrnula dvě kohorty pacientů s RR RS v Kanadě, USA, Rusku a v devíti evropských státech, v první bylo 188 pacientů rovnoměrně rozdělených do skupin s dávkami 10 mg, 2 mg, 0,5 mg a do skupiny s placebem, po třech měsících byla vyhodnocena interim analýza a na jejím základě vytvořena druhá kohorta 109 pacientů s dávkováním 1,25 mg, 0,25 mg a placebo v poměru 4 : 4 : 1. Pacienti byli ve věku 18‒55 let, měli nejméně jeden relaps RS v roce před vstupem do studie, dva relapsy v posledních dvou letech nebo jednu či více gadolinium enhancujících lézí na magnetické rezonanci (MR) v době screeningu do studie. Jejich EDSS (Expanded Disability Status Scale, hodnocení neurologického nálezu a invalidity) činilo 0‒5,0. V první kohortě byli pacienti léčeni plnou dávkou od randomizační návštěvy, ve druhé kohortě byla provedena maskovaná titrace dávky po schválení dodatku protokolu.
Primárním cílem studie bylo zjištění, zda efekt léku je závislý na dávce a na nalezení dávky omezující tvorbu lézí na MR během prvních tří měsíců léčby. Bylo použito inovativní adaptivní uspořádání studie se statistickým modelováním, protože u léků, jako jsou modulátory S1P receptorů, může se zvyšující se dávkou stoupat množství nežádoucích účinků. Vyšetření MR bylo prováděno jednou měsíčně (T1 vážené obrazy s gadoliniem, T2 vážené obrazy) a zjišťoval se parametr CUAL (combined unique active lesions, tedy součet gadolinium enhancujících lézí a nových nebo zvětšujících se T2 lézí, bez dvojího počítání ‒ tedy nová léze, která se objevila na T1 i na T2, byla počítána pouze jednou).
Sekundárními cílovými ukazateli byly měsíční počet CUAL na MR, počet pacientů bez MR aktivity, roční počet relapsů, počet pacientů bez relapsu.
Při vstupu do studie mělo 44‒57 % pacientů v každé skupině alespoň jednu gadolinium enhancující lézi, šlo tedy o pacienty s vysokou aktivitou nemoci. Skóre EDSS bylo mezi 2,0 a 2,4, doba trvání nemoci činila v průměru 6–8,7 roku. Celkem 79 % pacientů v první kohortě a 95 % v druhé kohortě dokončilo studii. Hodnocení ukázalo významnou závislost mezi dávkou léku a efektem na CUAL během tří měsíců podávání. Počet CUAL byl redukován o 82 % u dávky 10 mg, o 72 % u dávky 2 mg, o 66 % u dávky 1,25 mg, o 50 % u dávky 0,5 mg a o 35 % u dávky 0,25 mg. Odpověď na dávku zůstala konzistentní i po šesti měsících. Ačkoliv studie nebyla uspořádána ke zjištění efektu na výskyt relapsů, došlo k významné redukci počtu relapsů u dávky 2 mg v porovnání s placebem. Nežádoucích účinků v první kohortě, zaměřené i na bezpečnost, bylo více než u placeba. Nejčastěji šlo o bolesti hlavy, bradykardii, závratě a nazofaryngitidu. Nejzávažnějším nežádoucím účinkem byl atrioventrikulární blok II. stupně ve skupině léčené 2 mg siponimodu. Titrace dávky ve druhé kohortě výrazně omezila bradykardii spojenou s první dávkou modulátoru S1P receptorů. Vyšší hodnoty jaterních testů byly častější u skupin léčených 2 mg a 10 mg siponimodu. Jeden pacient s anamnézou uveitidy vyvinul makulární edém. Ve studii se nevyskytly žádné závažné ani oportunní infekce. Dávka siponimodu 2 mg měla efekt velmi podobný dávce 10 mg, u níž bylo pozorováno nejvíce nežádoucích účinků.
Studie
EXPAND
V roce 2018 byla publikována randomizovaná, dvojitě zaslepená studie fáze III se siponimodem (EXPAND) použitým k léčbě SP RS [11]. Tato multicentrická studie probíhala ve 292 centrech ve 31 zemích. Do studie byli zahrnuti pacienti ve věku 18‒60 let s EDSS 3,0‒6,5 s původně RR formou nemoci podle McDonaldových kritérií 2010, s prokázanou progresí stavu za poslední dva roky a bez známek relapsu v posledních třech měsících. Základní část studie byla zaslepená a randomizovaná, nyní probíhá následná pokračující otevřená fáze s plánem sbírat data o účinnosti a bezpečnosti celkově po dobu 10 let. Vylučovacími kritérii byly jakákoliv závažná choroba imunitního, kardiálního a plicního systému, přítomnost makulárního edému, nekontrolovaný diabetes, genotyp CYP2C9*3*3 a negativní protilátky proti viru varicella zoster.
Pacienti byli randomizováni v poměru 2 : 1 k podávání siponimodu nebo placeba. V prvních šesti dnech probíhala titrace dávky od 0,25 mg do 2 mg udržovací dávky. Retitrace byla nutná, pokud došlo k přerušení léčby na čtyři či více dnů. Snížení tepové frekvence a počtu lymfocytů jsou známé farmakologické účinky siponimodu, které mohou odslepit studijní subjekty. Proto pro titraci byl ve studijním týmu přítomen další lékař a laboratorní výsledky nebyly zasílány do centra, pokud v nich nebyly závažné abnormality. Každé tři měsíce bylo prováděno neurologické vyšetření (EDSS). Vyšetření MR bylo provedeno každých 12 měsíců a na konci zaslepené fáze studie, snímky byly hodnoceny v jednom čtecím centru, NeuroRX Research, Montreal, QC, Kanada. Pacienti s potvrzenou progresí invalidity trvající šest měsíců podepisovali nový informovaný souhlas s léčbou a měli možnost zůstat ve studii za stávajícího zaslepení nebo přejít k otevřené léčbě siponimodem či zůstat pouze sledováni, neléčeni nebo léčeni jiným lékem.
Primárním cílovým ukazatelem studie byla doba do potvrzené tři měsíce trvající progrese disability (confirmed disability progression, CDP). Ta byla definována jako zvýšení EDSS o jeden stupeň, jestliže vstupní skóre EDSS bylo 3,0‒5,0, nebo o půl stupně EDSS, bylo li vstupní skóre 5,5‒6,5. Konfirmace proběhla při další vizitě za tři měsíce. Dva hlavní sekundární cílové ukazatele představovaly: zhoršení (potvrzené po třech měsících) v testu chůze na čas (T25FWT, test rychlé chůze na 25 stop) a změna objemu hyperintenzních lézí na MR v T2 vážených obrazech od začátku studie. Hodnocení mělo ještě řadu dalších sekundárních cílových ukazatelů. Doba do progrese disability potvrzené po třech měsících byla analyzována v podskupinách pacientů předdefinovaných podle přítomnosti nebo nepřítomnosti relapsů v posledních dvou letech před randomizací, rychlé progrese (zvýšení EDSS o alespoň 1,5 bodu v posledních dvou letech před randomizací) a podle MSSS (Multiple Sclerosis Severity Score) 4 či více při zahájení studie. Statistická analýza předpokládala, že pozorování 374 událostí progrese disability potvrzené po třech měsících dávalo studii 90% sílu detekovat 30% redukci rizika této progrese.
Screenováno bylo 2 092 pacientů, randomizováno bylo 1 651 pacientů, 1 105 k podávání siponimodu, 546 k podávání placeba, z toho 903 a 424 studii dokončilo. Medián času ve studii činil 21 měsíců.
Riziko tříměsíční CDP v porovnání s placebem bylo sníženo o 21 % (poměr rizik [HR] 0,79; p = 0,0134), graf 1A, B. Rozdíl nebyl pozorován ve výsledcích testu chůze T25FWT ani u pacientů se vstupním skóre EDSS 5,5 či méně. Riziko šes
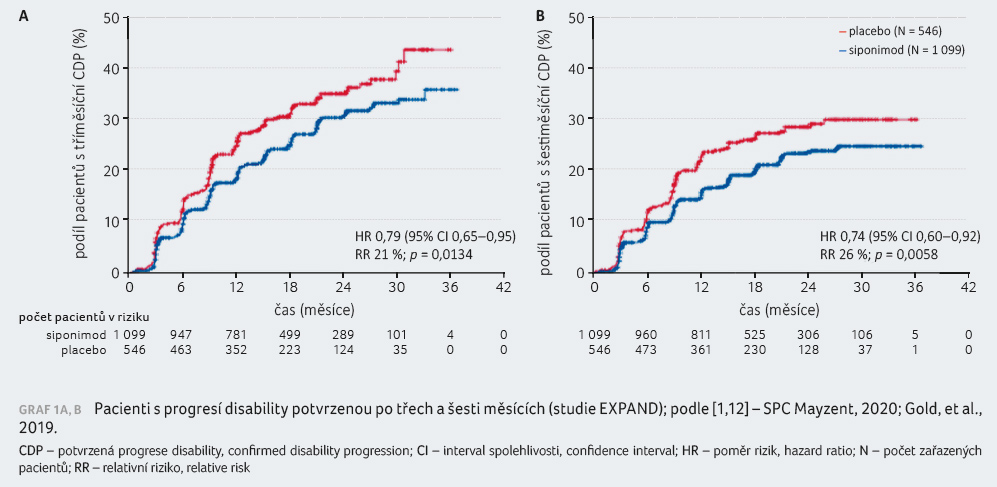
timěsíční CDP pak bylo redukováno siponimodem o 26 %. Vliv siponimodu na MR parametry byl v souladu s klinickým efektem: objem T2 lézí narůstal pomaleji (p < 0,0001), objem mozku klesal pomaleji (p < 0,0001), 89 % pacientů léčených siponimodem nemělo žádné gadolinium enhancující léze po dobu studie (67 % pacientů s placebem), 57 % (siponimod) versus 37 % pacientů (placebo) nemělo žádné nové nebo zvětšující se T2 léze. V rámci exploratorních cílů byla hodnocena i změna rychlosti kognitivního zpracování podle Symbol Digit Modality Test (SDMT) skóre. Statisticky významné snížení rizika čtyřbodového zhoršení v SDMT činilo 25 % [1].
Snížení rizika progrese disability bylo potvrzeno i při analýze podskupiny pacientů (n = 779) s aktivním onemocněním (definovaných jako pacienti s relapsem během dvou let před vstupem do studie a/nebo přítomností T1 gadolinium enhancujících lézí na začátku studie). U této kategorie činila redukce rizika tříměsíční a šestiměsíční CDP 31 %, resp. 37 % oproti placebu [1,12].
Celkem 89 % pacientů léčených siponimodem a 82 % pacientů v placebové skupině hlásilo nežádoucí účinek, závažný nežádoucí účinek mělo 18 % pacientů léčených siponimodem a 15 % pacientů s placebem, 8 % pacientů v siponimodové skupině a 5 % pacientů v placebové skupině ukončilo studii pro nežádoucí účinek. Nejčastějšími nežádoucími účinky v obou léčebných ramenech byly bolesti hlavy, nazofaryngitida, zánět močových cest a pády. Hypertenze byla hlášena u 10 % pacientů ve skupině se siponimodem a u 8 % ve skupině s placebem. Závažné nežádoucí účinky hlášené alespoň 0,5 % pacientů v kterékoliv skupině představovaly zvýšené hodnoty jaterních testů, bazaliom, komoce, deprese, močová infekce, pokus o sebevraždu, poruchy chůze, relaps RS a paraparéza.
Proporčně více pacientů léčených siponimodem mělo nežádoucí účinky spojené s modulací S1P receptorů: bradykardii při zahájení léčby, hypertenzi, lymfopenii, makulární edém. Počty malignit byly podobné v obou léčebných ramenech. Stejně tak počty nežádoucích účinků a závažných nežádoucích účinků spojených s infekcemi byly podobné vyjma herpes zoster, který se objevil častěji u siponimodu (2 %) v porovnání s placebem (1 %). Ve skupině se siponimodem byla hlášena jedna zosterová meningitida.
Celkem 82 % pacientů bylo kontrolováno kardiální telemetrií prvních šest dnů. Byl zaznamenán pokles o maximálně 5,3 tepu za minutu (4 h po aplikaci dávky), u placeba o 1,2 tepu (1 h po aplikaci). Nebyl zaznamenán žádný případ atrioventrikulárního bloku II. stupně.
Studie zkoumala typickou populaci sekundárně progresivních pacientů, téměř dvě třetiny nemocných neměly v posledních dvou letech relaps, jen 20 % osob mělo fokální zánětlivou aktivitu na MR v podobě gadolinium enhancujících lézí a 50 % osob potřebovalo oporu při chůzi. Přesto v této populaci byl siponimod schopen oddálit další progresi měřenou jako CDP po třech měsících. S tím byly konzistentní i nález zpomalení tvorby nových ložisek na MR i zpomalení ztráty mozkové tkáně. Analýza podskupin ukázala, že efekt siponimodu je přítomen i v rámci těchto subpopulací, ale zmenšuje se s věkem a vstupní disabilitou, trváním nemoci a zmírňující se zánětlivou aktivitou. V porovnání s velmi účinným protizánětlivým lékem natalizumabem, který byl testován u sekundární progrese a neprokázal očekávaný efekt, se zdá, že siponimod má účinek nejen protizánětlivý, ale i na vlastní progresi onemocnění. Zvyšování EDSS, jak ho vidíme v sekundární progresi, má zásadní vliv na kvalitu života pacienta, jeho denní aktivity a soběstačnost. Podíváme li se na výsledky studie EXPAND, pětina (CDP po třech měsících) a čtvrtina (CDP po šesti měsících) klinicky relevantního zhoršení může být při léčbě siponimodem ušetřena.
Díky uspořádání umožňujícímu progredujícím pacientům přejít na účinnou léčbu již během zaslepeného hodnocení se jistě snížila statistická síla studie, která měla placebem kontrolovanou část kratší než ostatní podobná klinická sledování. Také možný vliv předchozí léčby remitujícího stadia choroby může být limitací této studie při analýze podskupin podle vstupní aktivity. Bezpečnostní profil léku se zásadně neodlišuje od ostatních léků skupiny modulátorů S1P receptorů. Titrace dávky snižuje nároky na monitoraci první dávky a umožňuje tak snazší začátek léčby pro pacienta i lékaře. Siponimod tedy pokrývá populaci pacientů, která zatím zůstává bez možnosti léčby založené na důkazech.
V České republice se studie EXPAND účastnilo pět RS center, zařazeno bylo 35 pacientů, z nichž 27 pokračuje v extenzi studie.
Léčba v klinické praxi je zatím umožněna pro specializovaná centra podle paragrafu 16.
Kontraindikace
Siponimod je kontraindikován u pacientů, kteří:
- jsou přecitlivělí na léčivou látku, arašídy, sóju nebo na kteroukoliv pomocnou látku uvedenou v souhrnu údajů o přípravku,
- trpí syndromem imunodeficience,
- mají v anamnéze progresivní multifokální leukoencefalopatii nebo kryptokokovou meningitidu,
- mají aktivní maligní onemocnění,
- mají těžké poškození jater (třída C podle Childa‒Pugha),
- v předchozích šesti měsících prodělali infarkt myokardu, nestabilní anginu pectoris, cévní mozkovou příhodu/tranzitorní ischemickou ataku, dekompenzované srdeční selhání (vyžadující hospitalizaci) nebo srdeční selhání třídy III/IV podle New York Heart Association (NYHA),
- mají v anamnéze atrioventrikulární blokádu druhého stupně typu Mobitz II, atrioventrikulární blokádu třetího stupně, sinoatriální srdeční blok nebo sick sinus syndrom, pokud nemají implantovaný kardiostimulátor,
- mají homozygotní genotyp CYP2C9*3 (CYP2C9*3*3) ‒ tzv. slabí metabolizéři,
- otěhotní nebo jsou ženami v plodném věku a nepoužívají účinnou antikoncepci.
Siponimod se dále nedoporučuje u pacientů, kteří mají těžkou srdeční arytmii vyžadující antiarytmika třídy Ia (chinidin, prokainamid) nebo III (amiodaron, sotalol), blokátory kalciových kanálů (např. verapamil, diltiazem) a další léky (např. ivabradin nebo digoxin), o nichž je známo, že snižují tepovou frekvenci, mají v anamnéze symptomatickou bradykardii nebo recidivující synkopy, nekontrolovanou hypertenzi nebo těžkou neléčenou spánkovou apnoi, mají prodloužení intervalu QTc > 500 ms.
Nežádoucí
účinky
K nejčastějším nežádoucím účinkům užívání léku patří bolest hlavy (15 %) a hypertenze (12,6 %). Ta byla v klinické studii fáze III u pacientů se SP RS častěji hlášena u osob léčených siponimodem (12,6 %) než u nemocných s placebem (9,0 %). Léčba byla spojena se vzestupem systolického a diastolického krevního tlaku projevujícím se krátce po zahájení léčby a dosahujícím maxima přibližně po šesti měsících léčby (systolický tlak o 3 mm Hg a diastolický tlak o 1,2 mm Hg) a následně stabilizovaným.
Celková incidence infekcí byla srovnatelná u pacientů léčených siponimodem a pacientů v placebové skupině (49,0 % vs. 49,1 %). Nicméně u siponimodu byla v porovnání s placebem hlášena vyšší četnost infekcí virem varicella zoster (2,5 % vs. 0,7 %). V extenzi klinické studie fáze III byl hlášen případ kryptokokové meningitidy.
U pacientů byl v porovnání s placebem hlášen vyšší výskyt makulárního edému (1,8 % vs. 0,2 %). Makulární edém se obecně zmírnil nebo spontánně vymizel po ukončení podávání přípravku. Zahájení léčby vede k přechodnému poklesu srdeční frekvence a může být také spojeno se zpomalením atrioventrikulárního vedení. Bradyarytmie byla hlášena u 6,2 % pacientů léčených siponimodem v porovnání s 3,1 % ve skupině placeba a atrioventrikulární blokády se objevily u 1,7 % pacientů léčených siponimodem v porovnání s 0,7 % ve skupině placeba.
U pacientů byly hlášeny zvýšené hodnoty jaterních enzymů (většinou zvýšení koncentrace alaninaminotransferázy, ALT). V klinické studii fáze III bylo zvýšení hodnot jaterních funkčních testů častěji pozorováno u pacientů léčených siponimodem (11,3 %) než u pacientů s placebem (3,1 %), zejména kvůli zvýšení hodnot jaterních aminotransferáz (ALT/aspartátaminotransferázy, AST) a gamaglutamyltransferázy. Zvýšení se většinou objevilo během prvních šesti měsíců po zahájení léčby. Koncentrace ALT se vrátily k normálu během přibližně jednoho měsíce po ukončení léčby siponimodem.
Lékové
interakce
Během zahájení léčby siponimodem nemají být užívána současně antiarytmika třídy Ia (např. chinidin, prokainamid) nebo třídy III (např. amiodaron, sotalol), přípravky prodlužující interval QT se známými arytmogenními vlastnostmi, blokátory kalciových kanálů snižující srdeční frekvenci (např. verapamil nebo diltiazem) nebo další látky, které mohou snižovat srdeční frekvenci (např. ivabradin nebo digoxin) kvůli možným aditivním účinkům na srdeční frekvenci. Současné použití těchto látek během zahájení léčby může být spojeno s těžkou bradykardií a srdeční blokádou.
Při zahajování léčby siponimodem u pacientů léčených betablokátory je nutná opatrnost kvůli aditivním účinkům na snížení srdeční frekvence. Léčba betablokátory může být zahájena u pacientů léčených stálou udržovací dávkou siponimodu.
Vakcinace
Použití živých atenuovaných vakcín může vést k riziku infekce, a je proto třeba se mu během léčby siponimodem a po dobu až čtyř týdnů po jejím ukončení vyhnout. Během léčby siponimodem a až čtyři týdny po ní může být vakcinace méně účinná. Účinnost vakcinace není považována za ohroženou, pokud je léčba siponimodem pozastavena v období jednoho týdne před očkováním a až do čtyř týdnů po očkování.
Inhibitory
CYP2C9 a CYP3A4
Vzhledem k významnému zvýšení expozice siponimodu se nedoporučuje současné užívání siponimodu a léčivých přípravků, které způsobují slabou inhibici CYP2C9 a slabou nebo silnou inhibici CYP3A4. Tento souběžný lékový režim se může skládat ze slabého duálního inhibitoru CYP2C9/CYP3A4 (např. flukonazol) nebo slabého inhibitoru CYP2C9 v kombinaci se samostatným slabým nebo silným inhibitorem CYP3A4.
Siponimod lze kombinovat s většinou typů induktorů CYP2C9 a CYP3A4. Nicméně s ohledem na očekávaný pokles expozice siponimodu je nutné zvážit vhodnost a možný přínos léčby, pokud je siponimod kombinován:
se silnými induktory CYP3A4/slabými induktory CYP2C9 (např. karbamazepin) u všech pacientů bez ohledu na genotyp,
se slabými induktory CYP3A4 (např. modafinil) u pacientů s genotypem CYP2C9*1*3 nebo *2*3.
Současné podání siponimodu v dávce 2 mg denně v přítomnosti dávky 600 mg rifampicinu (silný induktor CYP3A4 a slabý induktor CYP2C9) denně snížilo AUCtau,ss a cmax,ss siponimodu o 57 %, respektive o 45 % u subjektů s CY2C9*1*1.
Perorální
kontraceptiva
Současné podání siponimodu nemělo klinicky relevantní vliv na farmakokinetiku a farmakodynamiku kombinovaných perorálních kontraceptiv obsahujících etinylestradiol a levonorgestrel. Proto účinnost zkoumaných perorálních kontraceptiv zůstává při léčbě siponimodem zachována. Nebyly provedeny žádné interakční studie s perorálními kontraceptivy obsahujícími jiné gestageny, avšak vliv siponimodu na účinek perorálních kontraceptiv se neočekává.
Těhotenství
a kojení
Siponimod je kontraindikován u žen ve fertilním věku, které nepoužívají účinnou antikoncepci. Proto musí být před zahájením léčby žen ve fertilním věku k dispozici negativní výsledek těhotenského testu a pacientky mají být poučeny ohledně závažných rizik pro plod. Ženy ve fertilním věku musejí používat účinnou antikoncepci během léčby a po dobu nejméně deseti dnů od poslední dávky siponimodu.
Nejsou k dispozici žádné nebo existují jen omezené údaje týkající se užití siponimodu u těhotných žen.
Studie na zvířatech prokázaly siponimodem indukovanou embryotoxicitu a fetotoxicitu. Proto je siponimod kontraindikován v těhotenství. Pokud žena otěhotní během léčby, musí být podávání siponimodu ukončeno. Ošetřující lékař by měl pacientku edukovat ohledně rizika škodlivých účinků na plod spojených s léčbou a je nutno provést ultrasonografické vyšetření.
Není známo, zda jsou siponimod nebo jeho hlavní metabolity vylučovány do lidského mateřského mléka. Siponimod a jeho hlavní metabolity jsou vylučovány do mléka potkanů, lék tedy nemá být užíván během kojení.
Seznam použité literatury
- [1] SPC Mayzent. Dostupné na: https://www.ema.europa.eu/en/documents/product‑information/mayzent‑epar‑product‑information_cs.pdf. Datum revize textu 6. 4. 2020.
- [2] Sospedra M, Martin R. Immunology of multiple sclerosis. Semin Neurol 2016; 36: 115–127.
- [3] Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol 2012; 8: 647–656.
- [4] Goodman AD, Anadani N, Gerwitz L. Siponimod in the Treatment of Multiple Sclerosis Expert Opin Investig Drugs 2019; 28: 1051‒1057.
- [5] Havrdova E, Kobelt G, Berg J, et al; European Multiple Sclerosis Platform. New insights into the burden and costs of multiple sclerosis in Europe: Results of the Czech Republic. Mult Scler 2017; 23(2 Suppl): 41‒52.
- [6] Behrangi N, Fischbach F, Kipp M. Mechanism of Siponimod: Anti‑Inflammatory and Neuroprotective Mode of Action. Cells 2019; 8: 24.
- [7] Gentile A, Musella A, Bulitta S, et al. Siponimid (BAF312) prevents synaptic neurodegeneration in experimental multiple sclerosis. J Neuroinflamm 2016; 13: 207.
- [8] O’Sullivan C, Schubart A, Mir AK, Dev KK. The dual S1PR1/S1PR5 drub BAF312 (Siponimod) attenuates demylenation in organotypic slice cultures. J Neuroinflamm 2016; 13: 31.
- [9] SPC Gilenya (fingolimod). Dostupné na: https://www.ema.europa.eu/en/documents/product‑information/gilenya‑epar‑product‑information_cs.pdf. Datum revize textu 16. 12. 2019.
- [10] Selmaj K, Li DK, Hartung HP, et al. Siponimod for patients with relapsing‑remitting multiple sclerosis (BOLD): an adaptive, dose‑ranging, randomised, phase 2 study. Lancet Neurol 2013; 12: 756–767.
- [11] Kappos L, Bar‑Or A, Cree BAC, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double‑blind, randomised, phase 3 study [published correction appears in Lancet. 2018 Nov 17; 392: 2170]. Lancet 2018; 391: 1263‒1273.
- [12] Gold R, Kappos L, Bar‑Or A, et al. Efficacy of siponimod in secondary progressive multiple sclerosis patients with active disease: the EXPAND study subgroup analysis. Poster 750 presented at: 35th Congress of the European Committee for Treatment and Research in Multiple Sclerosis; September 11‒13, 2019; Stockholm, Sweden.