Profil pacienta léčeného siponimodem v běžné klinické praxi
Souhrn:
Halúsková S, Vališ M. Profil pacienta léčeného siponimodem v běžné klinické praxi. Remedia 2021; 31: 534–539.
Roztroušená skleróza (RS) je chronické zánětlivé autoimunitní a neurodegenerativní onemocnění centrálního nervového systému postihující zejména mladé dospělé. Terapie RS zaznamenala v posledních letech výrazný pokrok. Sekundárně progresivní forma RS (SP‑RS) představuje celosvětově druhou nejčastější formu RS. Zachytit konverzi do sekundární progrese je však v klinické praxi velmi obtížné kvůli velkým interindividuálním rozdílům v průběhu nemoci a také pro absenci přesných diagnostických nástrojů a validovaných biomarkerů. Z tohoto důvodu je diagnóza SP‑RS často stanovena až retrospektivně a opožděna zhruba o tři roky. Zatímco pro relabující‑remitující RS máme k dispozici několik schválených léčiv modifikujících průběh choroby, efektivní lék pro SP‑RS dlouho chyběl. Siponimod, selektivní modulátor sfingosin‑1‑fosfátových receptorů, je prvním perorálním přípravkem schváleným k léčbě SP‑RS, u kterého bylo prokázáno, že zpomaluje klinicky potvrzenou progresi. Účinnost a bezpečnost siponimodu byla testována v klinické studii fáze III EXPAND. Siponimod je indikován u dospělých pacientů se SP‑RS se známkami aktivity onemocnění, tj. s prokázaným relapsem a/nebo zánětlivou aktivitou doloženou zobrazovacími vyšetřeními. Před zahájením léčby siponimodem musí být u pacienta zjištěn genotyp CYP2C9.
Summary:
Haluskova S, Valis M. Profile of a patient treated with siponimod in routine clinical practice. Remedia 2021; 31: 534–539.
Multiple sclerosis (MS) is a chronic inflammatory autoimmune and neurodegenerative disease of the central nervous system affecting most commonly young adults. There has been tremendous progress in the treatment of MS over recent years. Secondary‑progressive MS (SP‑MS) is the second most common form of MS. Determining the conversion to SP‑MS remains a clinical challenge due to the heterogeneous course of the disease among patients and the absence of validated biomarkers and precise diagnostic tools. Therefore, the SP‑MS diagnosis is often established retrospectively and delayed for up to three years. While there are many disease‑modifying drugs indicated for relapsing‑remitting MS, effective treatment for SP‑MS was lacking. Siponimod, a selective modulator of sphingosine‑1‑phosphate receptors, is the first oral drug to treat SP‑MS that showed a statistically significant decrease in disability progression. The efficacy and safety of siponimod were evaluated in phase III clinical trial EXPAND. Siponimod is indicated for treating adult SP‑MS patients with active disease evidenced by relapses or imaging features of inflammatory activity. The CYP2C9 genotype of the patient should be determined prior to initiating siponimod treatment.
Key words: multiple sclerosis, secondary progression, therapy, siponimod
Úvod
Roztroušená skleróza (RS) je chronické zánětlivé demyelinizační a neurodegenerativní onemocnění centrálního nervového systému (CNS), které postihuje především mladé dospělé a je asociováno s vysokou mírou invalidizace osob v produktivním věku [1]. Jedná se o multifaktoriální onemocnění, v jehož patogenezi sehrává klíčovou úlohu autoimunitní imunopatologická reaktivita a předpokládá se vliv faktorů jak genetických, tak environmentálních. Nemoc je charakterizována infiltrací leukocytů do CNS, lokální destrukcí myelinových obalů nervových vláken a postupnou ztrátou oligodendrocytů a axonů [2]. K úbytku mozkové tkáně dochází v tomto případě rychleji než při přirozeném procesu stárnutí organismu, což vede k nevratnému nárůstu neurologického postižení [3−5]. Onemocnění začíná u 85 % případů relabujícím remitujícím stadiem, kdy se ataky neurologických příznaků střídají s remisemi. Délka remise je individuální, k dalšímu relapsu může dojít za půl roku nebo za deset let. Po překročení určité míry poškození nervové tkáně se neurologický deficit stává trvalým a pozvolna progredujícím, dochází k vyčerpání rezerv CNS a k přechodu do sekundárně progresivní fáze. Podle údajů v literatuře se až u 80 % neléčených pacientů s relabující remitující RS (RR RS) rozvíjí přibližně po 15–20 letech trvání nemoci sekundárně progresivní forma RS (SP RS) [6,7]. Terapií je ale možné tento přechod zcela zásadně ovlivnit. Studie EPIC (multiple sclerosis Expression/genomics, Proteomics, Imaging, and Clinical study) doložila, že při aktivní léčbě konvertovalo do SP RS po 10 letech 6,4 % pacientů, po 20 letech 24,2 % nemocných [8].
Léčba RS zaznamenala v posledních dvou dekádách významný pokrok. Díky detailnějšímu a hlubšímu pochopení patogenetických mechanismů podílejících se na vzniku nemoci jsme v současnosti svědky výrazné akcelerace vývoje nových léčiv a jejich postupného zavádění do klinické praxe. Zatímco v 90. letech minulého století se výzkum zaměřoval především na RR RS, nyní je značná pozornost věnována progresivním formám nemoci, které jsou zkoumány stále intenzivněji. Dnes již máme k dispozici první léčivé přípravky pro terapii jak primárně progresivní formy RS (PP RS), tak i SP RS.
Progrese onemocnění a její
sledování v klinické praxi
V roce 2014 publikoval profesor
Lublin se spolupracovníky návrh nové klasifikace klinického
průběhu RS s cílem zdůraznit léčebně ovlivnitelné
zánětlivé změny oproti terapeuticky hůře ovlivnitelným změnám
neurodegenerativním. Podle tohoto nového třídění přistupujeme
k PP RS a SP RS souhrnně jako k progresivní
RS, která může být dále členěna na kategorie aktivní
nebo neaktivní RS – podle přítomnosti relapsu a/nebo nové
aktivity zaznamenané pomocí magnetické rezonance (MRI), stejně
jako s progresí či bez progrese – hodnoceno na základě
klinického vyšetření a objektivní míry změny v čase.
Progresivní onemocnění tak může být aktivní s progresí,
aktivní bez klinické progrese, neaktivní, ale s klinickou
progresí, resp. se může jednat o stabilní nemoc, tedy
neaktivní a bez progrese [9] (obr. 1).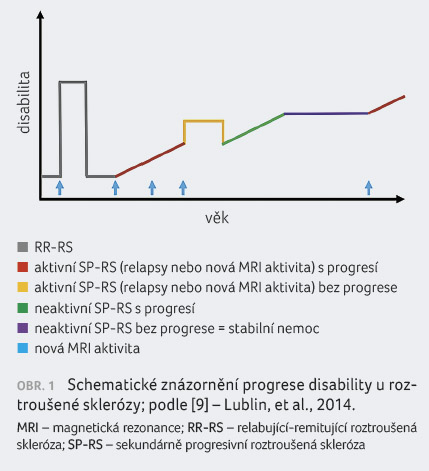
Předpokládá se, že první změny imunitních reakcí a patologické procesy v CNS mohou předcházet vlastní klinické manifestaci i o řadu let [10,11]. Bylo doloženo, že progrese začíná již od počátku onemocnění a postupuje pomalu, bez závislosti na klinických relapsech [12]. Běžná praxe potvrzuje, že vedle zhoršení po relapsu v podobě reziduálního neurologického deficitu (relapse associated worsening, RAW) vnímá řada nemocných určité trvalé zhoršování stavu bez zaznamenané ataky. Tuto skutečnost potvrdila i observační studie TOP (Tysabri Observational Program), která analyzovala celkem 5 562 pacientů s RR RS. Z výsledků vyplynulo, že zhruba 66 % celkového potvrzeného zhoršení disability (confirmed disability progression, CDP) představovala tzv. progrese nezávislá na relapsech (progression independent of relapse activity, PIRA) neboli „tichá progrese“ [13].
V praxi nejpoužívanější škálou k hodnocení vývoje disability u pacientů s RS je Kurtzkeho stupnice postižení, známá jako EDSS (Expanded Disability Status Scale). Její nevýhodou však je značná variabilita výsledků mezi vyšetřujícími a nedostatečná senzitivita k vyjádření skutečné disability pacientů s RS – horšící se klinický obraz se v celkovém kontextu nemusí promítnout do změny skóre. Mezi další užitečné a přesnější nástroje, které jsou široce využívány zejména v klinických hodnoceních, patří test chůze na 25 stop (Timed 25 foot Walk Test, T25WT), test jemné motoriky horních končetin (9 Hole Peg Test, 9HPT) a test zaměřený na stav kognitivních funkcí kvantifikující rychlost zpracování informací (Symbol Digit Modalities Test, SDMT). Tento test má sice vysokou senzitivitu pro diagnostiku poklesu kognitivních funkcí, v případě pacienta s vyšší kognitivní rezervou se však deficit nemusí odhalit hned při prvním vyšetření. Ukazuje se, že zhoršení kognice často i o roky předchází zhoršování fyzické disability kvantifikované pomocí EDSS a její hodnocení proto může posloužit jako důležitý indikátor progrese onemocnění.
Kromě klinicky měřitelných parametrů je možno progresi nemoci hodnotit i pomocí paraklinických vyšetřovacích metod. Jednu z nich představuje stanovení hodnot lehkých řetězců neurofilament v likvoru a séru, jejichž koncentrace odrážejí axonální poškození a korelují s aktivitou onemocnění, progresí disability i ztrátou objemu mozkové tkáně. Zatím převážně experimentální, neinvazivní metodou je optická koherenční tomografie (OCT), která kvantifikuje postižení sítnice a zrakového nervu a jeví se jako modalita jak diagnostická, tak prognostická. Ukázalo se, že nálezy na OCT korelují se změnou zrakové ostrosti, s atrofií mozku a kognitivními změnami u pacientů s RS. V praxi běžně používaná MRI hraje v managementu RS významnou roli – je zásadní při stanovení diagnózy, lze díky ní sledovat odpověď na léčbu a má také prediktivní význam. Jde o nejdůležitější a zatím jediný spolehlivý biomarker. Ani moderní sofistikované metody a relevantní funkční testy však nemusejí zcela zachytit drobné změny v čase, které vnímá sám pacient. Kontakt lékaře s pacientem probíhá v určitých pravidelných intervalech a je časově omezen, o to větší pozornost je potřeba věnovat udávaným subjektivním obtížím a emocím nemocného, jež vypovídají o zhoršené kvalitě života a naznačují skrytou progresi.
Zhoršování klinického stavu probíhá individuální rychlostí a závisí rovněž na podané léčbě. Není pochyb o tom, že při včasném zahájení odpovídající terapie se riziko rychlé progrese ireverzibilní disability výrazně snižuje. Italští autoři ve své práci zjistili, že pravděpodobnost dosažení hodnoty EDSS 6 v 55 letech byla u pacientů s RS přibližně stejná do roku 2000. U nemocných diagnostikovaných mezi lety 2001–2005 se pravděpodobnost dosažení EDSS 6 snížila o 37 % a v letech 2006–2010 dokonce o 46 % [14]. Tento výsledek úzce souvisí s dobovými terapeutickými možnostmi a reflektuje nejen urychlení diagnostického procesu s časnější iniciací terapie léky modifikujícími průběh onemocnění (disease modifying drugs, DMDs), ale také zavádění moderních, účinnějších přípravků na trh.
Sekundárně progresivní RS
Sekundárně progresivní RS představuje celosvětově druhou nejčastější formu RS [15]. Zachytit konverzi do sekundární progrese je však v klinické praxi velmi obtížné z důvodu neexistence přesné hranice mezi RR RS a SP RS [16]. Průběh onemocnění je interindividuálně značně heterogenní a dosud neexistují jasná klinická, zobrazovací, imunologická kritéria či validované biomarkery pro určení bodu přechodu. Stanovení diagnózy je proto problematické, často intuitivní a v naprosté většině případů až retrospektivní na základě anamnézy kontinuálního zhoršování neurologických funkcí u pacientů s vyšší mírou disability dle EDSS po počátečním relabujícím průběhu choroby [17]. Mezi nepříznivé prognostické faktory dřívější konverze do SP RS řadíme delší dobu trvání onemocnění, vyšší věk, infratentoriální lokalizaci lézí, výraznější atrofii mozkové tkáně, kouření, nízké sérové koncentrace vitaminu D, multifokální klinickou manifestaci včetně míšní symptomatiky, vyšší počet relapsů v počátcích nemoci, neúplné zotavení z první ataky a mužské pohlaví [18]. Vzhledem k diagnostickým obtížím je diagnóza často opožděna a bylo doloženo, že od první návštěvy, kdy lékař pojme podezření na progresi, do první návštěvy s jasnou diagnózou SP RS uběhnou zhruba tři roky. Období tzv. diagnostické nejistoty bylo popsáno až u 70 % nemocných [16]. Kromě absence spolehlivých diagnostických nástrojů diskutovaných výše byl důvodem oddalování diagnózy SP RS nedostatek odpovídajících terapeutických možností a s tím spojená psychická zátěž pacienta [19]. Pro pacienty s jistou diagnózou SP RS bývá typická zhoršená schopnost chůze s potřebou jedno /oboustranné opory, vysilující patologická únava, poruchy vyjadřování, paměti, deprese, sfinkterová dysfunkce, omezení sociálních kontaktů, nemocní jsou zvýšeně závislí na péči partnerů a rodiny, zaměstnání obvykle zvládají pouze na částečný úvazek. Oznámení diagnózy SP RS pro ně až donedávna znamenalo ukončení stávající léčby, strach z budoucnosti a pocity beznaděje ze skutečnosti, že jejich deficit se bude patrně už jen zhoršovat.
Obecně je SP RS charakterizována postupně narůstající disabilitou bez tendence k úpravě. Detailní definice koncipovaná na základě výsledů rozsáhlé studie MSBase (data 17 356 pacientů ze 113 RS center ve 34 zemích) s cílem časnější identifikace sekundární progrese a objektivizace kritérií definuje SP RS jako: progresi disability o 1 stupeň EDSS u pacientů s EDSS ≤ 5,5 nebo o 0,5 stupně při EDSS ≥ 6,0 bez přítomnosti relapsů, s minimální hodnotou EDSS 4,0 a postižením pyramidového funkčního systému dosahujícím minimálně stupně 2. Nutností je konfirmovaná progrese disability za tři měsíce včetně postižení pyramidových funkcí. Uvedená definice prokázala 87% přesnost ve srovnání s konsenzuálním stanoviskem nezávislých hodnotitelů a umožnila stanovit diagnózu SP RS o tři roky dříve, než by ji mohl vyslovit ošetřující lékař [20]. I když je tato definice v současnosti považována za nejspolehlivější a nejpraktičtější, je vhodná spíše pro klinická hodnocení, pro uplatnění v běžné praxi méně. Diskutovanou zůstává zejména hodnota EDSS minimálně 4,0, neboť se zdá, že u některých pacientů začíná sekundární progrese dříve a její nástup při nízkých hodnotách EDSS není raritní. Výsledky výzkumů ukazují, že u 70 % pacientů je stanovena diagnóza SP RS až při EDSS ≥ 6, což je poměrně závažná úroveň postižení vyžadující jednostrannou oporu při chůzi. Ovšem 76 % pacientů má rozpoznatelnou progresi při EDSS ≤ 3,0 a 84,8 % pacientů má nástup progrese již při EDSS ≤ 2 [16,21]. U pacientů s RR RS léčených DMD došlo k progresi do fáze SP RS v průměru už ve věku 38 let [22].
Původní teorie o přítomnosti zánětlivých procesů v časných fázích onemocnění a neurodegenerace až ve stadiích pokročilejších byly na základě nejnovějších vědeckých poznatků překonány. Dnes víme, že převažující zánět postupně přechází do neurodegenerativní fáze, nicméně jednotlivé pochody probíhají paralelně a liší se spíše kvantitou než kvalitou [23]. Platí, že již v čase diagnózy je možné nalézt známky neurodegenerace, na druhou stranu i po letech od počátku nemoci lze na okraji lézí zaznamenat zánět. Oproti časné fázi RS, kdy je porušena hematoencefalická bariéra (HEB) a aktivované imunitní buňky migrují z periferní krve do centrálního kompartmentu, dochází v progresivním stadiu k uzavření chronického zánětu za HEB, a z tohoto důvodu jsou možnosti terapeutické intervence výrazně omezené [24−26]. Po dlouhou dobu představovala SP RS velmi nepříjemné téma, protože jediné, co mohli lékaři pacientům v sekundární progresi ještě donedávna nabídnout, byla symptomatická terapie a intenzivní rehabilitace. Situace se však změnila s příchodem siponimodu – historicky prvního a zatím jediného schváleného léku v indikaci SP RS, u kterého bylo prokázáno, že zpomaluje klinicky potvrzenou progresi.
Siponimod a jeho (kontra)indikace
Schválení prvního perorálního DMD pro dospělé pacienty s aktivní SP RS v zemích Evropské unie proběhlo v roce 2020, v České republice je siponimod hrazen ze zdravotního pojištění od 1. února 2021. Registrace přípravku se opírá o výsledky rozsáhlé (celkem 1 645 pacientů z 292 RS center ve 31 zemích) randomizované, dvojitě zaslepené studie fáze III publikované v roce 2018. Klinická studie EXPAND srovnávala účinnost a bezpečnost siponimodu oproti placebu u typické populace pacientů se SP RS, tj. starších nemocných s delším trváním nemoci a vyšším stupněm disability. V rámci celého analyzovaného souboru vedla léčba siponimodem v porovnání s placebem k jednoznačnému snížení rizika tříměsíční i šestiměsíční CDP (o 21 %, resp. 26 %), ke statisticky signifikantnímu poklesu anualizované míry relapsů (o 55 % oproti placebu) i ke zpomalení procesu atrofizace mozku [27]. Ještě výraznější byl efekt siponimodu v subpopulaci pacientů s aktivní SP RS (přibližně polovina hodnocené populace), tedy u nemocných s dokumentovaným relapsem v průběhu dvou let před vstupem do studie a/nebo s minimálně jednou prokázanou gadolinium enhancující T1 lézí při screeningu. V tomto případě činila redukce rizika CDP po třech a šesti měsících 31 %, resp. 37 % oproti placebu a roční četnost relapsů byla redukována o 46 % v porovnání s neléčenými pacienty [28]. Přínos siponimodu byl průkazný také při hodnocení několika dalších, sekundárních cílových ukazatelů a rovněž bezpečnostní profil léku se ukázal jako poměrně příznivý [27]. Setrvalou účinnost přípravku potvrzují pětiletá data [29]. V rámci post hoc analýzy studie EXPAND bylo zjištěno, že siponimod oddaluje dobu do nutnosti použití invalidního vozíku – siponimod v porovnání s placebem prodloužil v celkové populaci střední čas do hodnoty EDSS ≥ 7 o více než čtyři roky [30].
Siponimod je selektivní modulátor sfingosin 1 fosfátových receptorů, konkrétně podtypů S1P1 a S1P5 exprimovaných na buňkách imunitního systému a CNS. Ve vztahu k receptoru S1P1 inhibuje výstup lymfocytů z lymfatické tkáně a omezuje tak jejich schopnost pronikat do CNS. Navíc jako lipofilní substance siponimod poměrně snadno přestupuje HEB a přímo v CNS tak ovlivňuje zánětlivé procesy probíhající při SP RS. Preklinická data naznačila i možné neuroprotektivní účinky podporující remyelinizaci a omezující synaptickou neurodegeneraci [31−33].
Na základě diskusí mezi
odborníky zabývajícími se RS byl stanoven optimální profil
pacienta se SP RS vhodného k léčbě siponimodem (obr.
2). Podle aktuálně platných indikačních kritérií lze
siponimod indikovat u dospělých pacientů se SP RS se
známkami aktivity onemocnění, tj. s prokázaným relapsem
během posledních dvou let a/nebo se zánětlivou aktivitou
na MRI mozku zahrnující gadolinium enhancující T1 léze
nebo nová a/nebo zvětšující se T2 ložiska. Pacient musí mít
vstupní hodnotu EDSS 4,0–6,5 a prokázanou progresi
disability o ≥ 1 stupeň při vstupním skóre EDSS < 5,5
či o 0,5 stupně při vstupní hodnotě EDSS > 5,5,
přičemž progrese disability trvá šest měsíců nezávisle
na relapsech [34]. V reálné praxi se tedy jedná
o pacienta s dosavadní RR RS léčeného DMD I. nebo
vyšší linie či bez biologické léčby, u něhož dochází
k postupné progresi disability bez přítomnosti relapsů,
s patrným zhoršováním kognitivních funkcí (pomalejší
zpracování informací, poruchy soustředění), narůstající
únavou, zhoršující se dysfunkcí močového měchýře nebo
s potížemi s rovnováhou včetně opakovaných pádů,
typická je rovněž vyšší míra hospitalizací. Pokud při léčbě
siponimodem dojde k dosažení EDSS ≥ 7 a ke ztrátě
schopnosti chůze, přípravek není dále hrazen. V případě,
že při podávání siponimodu přetrvává aktivita onemocnění
s více než jedním dokumentovaným relapsem, existuje možnost
přehodnocení průběhu nemoci s případným převedením
pacienta na léčbu přípravkem eskalační linie léčby
RR RS, který dosud nebyl podáván.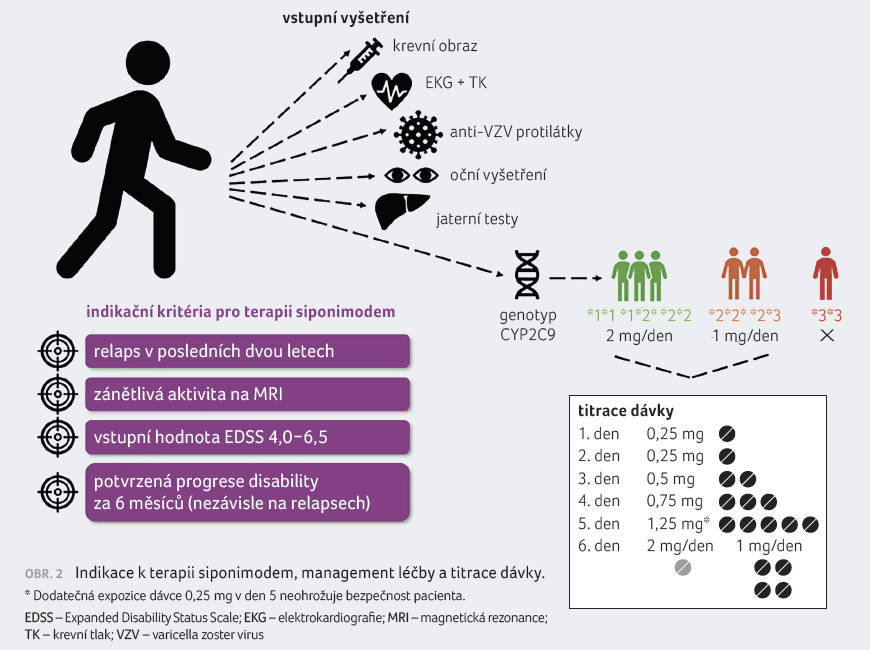
Před zahájením léčby je potřeba dodržet iniciační protokol (obr. 2): je nutné provést genotypizaci pro enzym cytochromu P450 2C9 (CYP2C9) z důvodu zjištění typu metabolizéra a určení vhodné dávky léku. Určité genotypové varianty totiž metabolizují siponimod s nižší účinností a rizikem několikanásobně vyšší expozice léku. Subjekty s CYP2C9*1*1, *1*2 a *2*2 se chovají jako silní metabolizéři (83–91 % populace) s cílovou dávkou léčiva 2 mg/den, subjekty s CYP2C9*1*3 a *2*3 jako středně silní metabolizéři s doporučenou poloviční dávkou léku a nositelé CYP2C9*3*3 by jako slabí metabolizéři (< 0,5 %) neměli siponimod užívat vůbec. Genetické testování polymorfismu CYP2C9 se provádí jednoduše z jednoho odběru krve, jelikož se ale jedná o vyšetření genetického materiálu, je vyžadován informovaný souhlas podepsaný pacientem. Dále vyšetřujeme krevní obraz včetně diferenciálního rozpočtu (absolutní hodnoty lymfocytů iniciálně ≥ 0,8 × 109), jaterní funkce, koncentraci protilátek proti viru varicella zoster (s doporučením vakcinace, pokud není imunoprotektivní hodnota), provádíme rovněž oční vyšetření (vyloučení makulárního edému), EKG a kontrolujeme hodnoty krevního tlaku. Dávku je zpočátku nutné titrovat vzhledem k možnému ovlivnění srdeční frekvence. Obě udržovací dávky (2 mg a 1 mg = 4× 0, 25 mg jednou denně) mají společné titrační schéma zajišťující jejich bezpečné dosažení a snižující nároky na monitoraci po zahajovací dávce (obr. 2).
Mezi kontraindikace podávání siponimodu patří kromě hypersenzitivity na léčivou látku syndrom imunodeficience, progresivní multifokální leukoencefalopatie nebo kryptokoková meningitida v anamnéze, aktivní nádorové onemocnění či těžká porucha funkce jater. Siponimodem nesmějí být léčeni ani pacienti s atrioventrikulární blokádou II. či vyššího stupně, sinoatriální blokádou, sick sinus syndromem (pokud nemají implantovaný kardiostimulátor) a lék nesmí být rovněž preskribován u jedinců, kteří prodělali v posledních šesti měsících infarkt myokardu, nestabilní anginu pectoris, iktus/tranzitorní ischemickou ataku a dekompenzované srdeční selhání nebo srdeční selhání třídy III/IV dle NYHA (New York Heart Association). Siponimod se nedoporučuje podávat souběžně s antiarytmiky třídy Ia a III, blokátory kalciových kanálů ani betablokátory, není li klidová srdeční frekvence > 50 tepů/min. Lék je kontraindikován u těhotných a kojících žen [34].
Závěr
Na RS bychom neměli nahlížet pouze klinickým pohledem, který se ve vztahu k probíhajícím patologickým procesům jeví jako opožděný a nedostatečný. I navzdory extenzivnímu výzkumu a snahám nalézt biomarkery, resp. vhodné kombinace biomarkerů, které by předpověděly vývoj nemoci u konkrétního pacienta v delším časovém horizontu, zatím bohužel neumíme predikovat, u koho se bude onemocnění vyvíjet příznivým či nepříznivým způsobem. Jediným správným principem a terapeutickým imperativem je tedy působit preventivně – potlačit probíhající zánět nasazením vhodné biologické léčby ihned po stanovení definitivní diagnózy RR RS, pokusit se co nejvíce eliminovat negativní vlivy životního stylu, které se na progresi onemocnění podílejí, a tím oddálit přechod do progresivní fáze. Pokud ale pacient do SP RS dospěje, zásadní je zkrátit období diagnostické nejistoty na minimum a toto stadium včas rozpoznat. Sekundární progrese však zůstává i nadále velkou diagnostickou výzvou, a to nejen proto, že mnohdy probíhá skrytě. Důležitým faktorem je rovněž stárnutí a jeho vliv na zhoršující se RS. U starších nemocných je klinický obraz ovlivněn přítomnými, zejména cévními komorbiditami, různými infekčními komplikacemi, případně progredujícími psychopatologiemi. V současnosti, kdy máme k dispozici specifickou terapii SP RS, je namístě využít „okno příležitosti“ a snažit se průběh nemoci i v této fázi co nejvíce zpomalit. Z pohledu pacientů, kteří si chtějí udržet nezávislost co nejdéle, je zpomalení progrese nemoci nesmírně důležité. Naději pro tuto skupinu pacientů přináší siponimod, s jehož pomocí, doufáme, budou moci tohoto cíle dosáhnout.
Seznam použité literatury
- [1] Oh J, Vidal‑Jordana A, Montalban X. Multiple sclerosis: clinical aspects. Curr Opin Neurol 2018; 31: 752–759.
- [2] Arneth BM. Impact of B cells to the pathophysiology of multiple sclerosis. J Neuroinflammation 2019; 16: 128.
- [3] Pandit L. No Evidence of Disease Activity (NEDA) in Multiple sclerosis – shifting the goal posts. Ann Indian Acad Neurol 2019; 22: 261–263.
- [4] Schippling S, Ostwaldt AC, Suppa P, et al. Global and regional annual brain volume loss rates in physiological aging. J Neurol 2017; 264: 520–528.
- [5] De Stefano N, Stromillo ML, Giorgio A, et al. Establishing pathological cut‑offs of brain atrophy rates in multiple sclerosis. J Neurol Neurosurg Psychiatry 2016; 87: 93–99.
- [6] Brown JWL, Coles A, Horakova D, et al. Association of initial disease‑modifying therapy with later conversion to secondary progressive multiple sclerosis. JAMA 2019; 321: 175 –187.
- [7] Weinshenker BG, Bass B, Rice GP, et al. The natural history of multiple sclerosis: a geographically based study, I: clinical course and disability. Brain 1989; 112 (Pt 1): 133– 146.
- [8] University of California, San Francisco MS‑EPIC Team:, Cree BA, Gourraud PA, Oksenberg JR, et al. Long‑term evolution of multiple sclerosis disability in the treatment era. Ann Neurol 2016; 80: 499–510.
- [9] Lublin FD, Reingold SC, Cohen JA, et al. Defining the clinical course of multiple sclerosis: the 2013 revisions. Neurology 2014; 83: 278–286.
- [10] Giovannoni G. The neurodegenerative prodrome in multiple sclerosis. Lancet Neurol 2017; 16: 413–414.
- [11] Novakova L, Axelsson M, Malmeström C, et al. Searching for neurodegeneration in multiple sclerosis at clinical onset: diagnostic value of biomarkers. PLoS One 2018; 13: e0194828.
- [12] University of California, San Francisco MS‑EPIC Team:, Cree BAC, Hollenbach JA, Bove R, et al. Silent progression in disease activity‑free relapsing multiple sclerosis. Ann Neurol 2019; 85: 653–666.
- [13] Kappos L, Butzkueven H, Wiendl H, et al. Greater sensitivity to multiple sclerosis disability worsening and progression events using a roving versus a fixed reference value in a prospective cohort study. Mult Scler 2018; 24: 963–973.
- [14] Capra R, Cordioli C, Rasia S, et al. Assessing long‑term prognosis improvement as a consequence of treatment pattern changes in MS. Mult Scler 2017; 23: 1757–1761.
- [15] Khurana V, Sharma H, Medin J, et al. Estimated prevalence of diagnosed secondary progressive multiple sclerosis in the Americas and Europe: results from a systematic literature search. Neurology 2018; 90(15 Suppl): P2.380.
- [16] Katz Sand I, Krieger S, Faqrrell C, et al. Diagnostic uncertainty during the transition to secondary progressive multiple sclerosis. Mult Scler 2014; 20: 1654–1657.
- [17] Oh J, Alikhani K, Bruno T, et al. Diagnosis and management of secondary‑progressive multiple sclerosis: time for change. Neurodegener Dis Manag 2019; 9: 301–317.
- [18] Larochelle C, Uphaus T, Prat A, et al. Secondary progression in multiple sclerosis: neuronal exhaustion or distinct pathology? Trends Neurosci 2016; 39: 325–339.
- [19] O'Loughlin E, Hourihan S, Chataway J, et al. The experience of transitioning from relapsing remitting to secondary progressive multiple sclerosis: views of patients and health professionals. Disabil Rehabil 2017; 39: 1821–1828.
- [20] Lorscheider J, Buzzard K, Jokubaitis V, et al. Defining secondary progressive multiple sclerosis. Brain 2016; 139 (Pt 9): 2395–2405.
- [21] Kremenchutzky M, Rice GP, Baskerville J, et al. The natural history of multiple sclerosis: a geographically based study 9: observations on the progressive phase of the disease. Brain 2006; 129 (Pt 3): 584–594.
- [22] Bsteh G, Ehling R, Lutterotti A, et al. Long term clinical prognostic factors in relapsing‑remitting multiple sclerosis: insights from a 10‑year observational study. PLoS One 2016; 11: e0158978.
- [23] Correale J, Marrodan M, Ysrraelit MC. Mechanisms of neurodegeneration and axonal dysfunction in progressive multiple sclerosis. Biomedicines 2019; 7: 14.
- [24] Sospedra M, Martin R. Immunology of multiple sclerosis. Semin Neurol 2016; 36: 115–127.
- [25] Lassmann H, van Horssen J, Mahad D. Progressive multiple sclerosis: pathology and pathogenesis. Nat Rev Neurol 2012; 8: 647–656.
- [26] Ontaneda D. Progressive multiple sclerosis. Continuum (Minneap Minn) 2019; 25: 736–752.
- [27] Kappos L, Bar‑Or A, Cree BAC, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double‑blind, randomised, phase 3 study. Lancet 2018; 391: 1263–1273.
- [28] Gold R, Kappos L, Bar‑Or A, et al. Efficacy of siponimod in secondary progressive multiple sclerosis patients with active disease: the EXPAND study subgroup analysis. ECTRIMS Online Library 2019; 279110: P750.
- [29] Giovannoni G, Kappos L, Fox R, et al. Sustained reduction of disability and cognitive decline with long‑term siponimod treatment in patients with active SPMS: EXPAND data up to 5 years. MSVirtual 2020; P0238.
- [30] Bar‑Or A, Buckle GJ, Cohan SL, et al. Exploring the safety and tolerability of conversion to siponimod in patients with relapsing forms of multiple sclerosis: design of the 6‑month prospective EXCHANGE study. ECTRIMS Online Library 2019; 278607: P1407.
- [31] Goodman D, Anadani N, Gerwitz L. Siponimod in the treatment of multiple sclerosis. Expert Opin Investig Drugs 2019; 28: 1051–1057.
- [32] Behrangi N, Fischbach F, Kipp M. Mechanism of siponimod: anti‑Inflammatory and neuroprotective mode of action. Cells 2019; 8: 24.
- [33] Martin E, Urban B, Beerli C. Siponimod (BAF312) is a potent promyelinating agent: preclinical mechanistic observations. ECTRIMS Online Library 2019; 278577: P1376.
- [34] SPC Mayzent. Dostupné na: https://www.ema.europa.eu/en/documents/product‑information/mayzent‑epar‑product‑information_en.pdf.