Role etnicity při léčbě inhibitory kináz v onkologii
Souhrn:
Jedním z faktorů ovlivňujících terapeutickou odpověď a toxicitu léčiv je etnicita. Ta se nemusí vždy opírat pouze o geneticky podmíněné odlišnosti mezi populacemi, ale může také vycházet z kulturních a socioekonomických rozdílů. Tyto vnější i vnitřní faktory mohou mít souvislost jak se změnami ve farmakodynamice, tak ve farmakokinetice. Příkladem léčiv, u kterých se vliv etnicity dostává do centra pozornosti, jsou inhibitory kináz. Ačkoliv se tato moderní cílená terapie začíná uplatňovat v řadě oblastí mimo onkologii, zůstává léčba nádorových onemocnění její hlavní doménou. V rámci klinického hodnocení byla většina zástupců této skupiny testována především u kavkazské a asijské populace. Širší zkušenosti s léčbou u ostatních populací dosud spíše chybějí. Následující text se pokusí sumarizovat závěry řady studií, které zkoumaly variabilitu v terapeutických odpovědích a/nebo v toxicitě inhibitorů kináz u různých etnik, a upozornit na významné rozdíly u vybraných skupin nádorových onemocnění (CML, NSCLC, RCC).
Summary:
Ethnicity is one of the factors that influence therapeutic response and drug toxicity. It is not based only on genetic variance between populations but also on cultural and socioeconomic differences. These outer and inner factors may be connected with differences both in pharmacodynamics and pharmacokinetics. Kinase inhibitors are examples of drugs for which ethnicity is becoming a center of attention. Although this modern targeted therapy is being used outside of the field of oncology more and more, the treatment of cancer remains its main domain. In the clinical trial setting, most of the drugs in this class were tested on Caucasian and Asian population. More complex experience with this treatment in other populations has been lacking up till now. The following text attempts to summarize the outcomes of many studies that assessed the variability in therapeutic responses and/or toxicity of kinase inhibitors in different ethnic groups and to point out important variance in selected groups of cancer diseases (CML, NSCLC, RCC).
Key words: targeted therapy, ethnicity, pharmacogenetics, tyrosine kinase inhibitors, oncology
Tak velký zájem jde ruku v ruce s neustále se prohlubujícími znalostmi lidského genomu. Ten v sobě skrývá genetickou informaci více než 500 proteinových kináz, v anglické literatuře je proto často používán pojem kinome [2]. Řada z těchto kináz je spojována se vznikem a s následnou progresí nádorového onemocnění. Výzkum v této oblasti probíhá již 40 let. První zmínky v literatuře pocházejí již z konce 70. let, kdy byla proteinkináza poprvé označena jako onkogen [3]. V roce 1981 bylo následně potvrzeno, že rostlinné estery forbolu, které způsobují hyperaktivaci proteinkinázy C (PKC), souvisejí s růstem nádorů [4]. V návaznosti na tento základní výzkum započala v následujících letech syntéza prvních molekul – inhibitorů proteinkináz. Ačkoliv se první látky potýkaly s nedostatečnou specificitou a účinností, došlo nakonec k uvedení prvního inhibitoru kináz na trh (imatinib). Jeho přínos znamenal v terapii CML určitý přelom [5,6]. Dnes je imatinib pro své „široké spektrum“ schválen v řadě dalších indikací − gastrointestinální stromální tumor (GIST), akutní lymfatická leukemie (AML), chronická eozinofilní leukemie, myelodysplastické/myeloproliferativní onemocnění, dermatofibrosarcoma protuberans.
Kinázy – cílové místo cílené terapie
Kinázy patří mezi enzymy, které se
podílejí na přenosu fosfátové skupiny z ATP
na protein, čímž dochází k jeho aktivaci. Opačnou
funkci, odebrání fosfátové skupiny z proteinu a jeho
inaktivaci, plní fosfatázy. Tyto vzájemně protichůdné reakce
modulují četné aktivity proteinů v buňce, často v reakci
na vnější stimul [7−9]. Tímto způsobem hrají kinázy
jednu z klíčových rolí v signalizaci nádorových
buněk, především pak v karcinogenezi a tvorbě
metastáz, neboť většina proteinkináz podporuje buněčnou
proliferaci, přežití a migraci buněk [10]. Jsou li
nadměrně exprimovány nebo je li jejich aktivita příliš
vysoká, jsou spojovány s onkogenezí [11]. Jak již bylo
zmíněno, dysregulace aktivit kináz je nicméně v posledních
letech spojována také s řadou onemocnění nespadajících
do oblasti onkologie. Dnes již jsou inhibitory kináz používány
také v pneumologii či v revmatologii. Z výzkumů
ale vyplývá, že význam kináz zasahuje také do dalších
oblastí − imunologie, neurologie, infektologie a dalších
[12−15].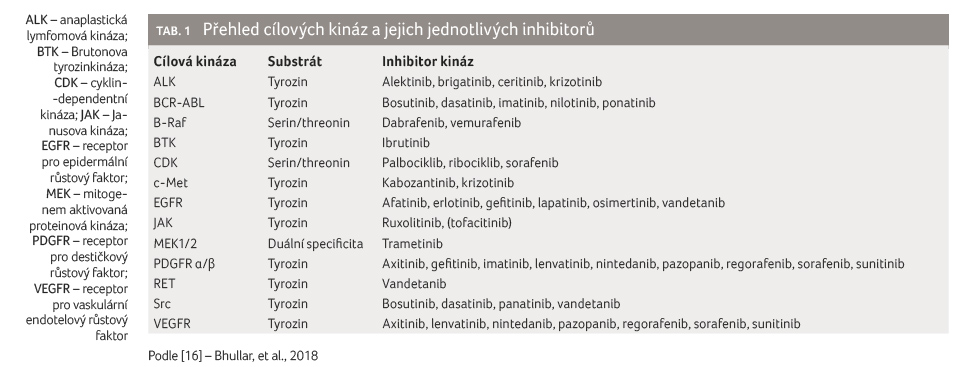
Z tabulky 1 [16] vyplývá, že převažujícím substrátem kináz, které jsou dnes ovlivnitelné dostupnými inhibitory, je tyrozin. Proto v následujícím textu bude v obecné rovině používána zkratka TKI (tyrozinkinázový inhibitor). Kromě kináz uvedených v tabulce 1 je pozornost v posledních letech zaměřena také na inhibitory Aurora kináz [17], kaseinové kinázy II (CKII) [18], fokální adhezní kinázy (FAK, PTK2) [19], proteinové kinázy B (PKB, Akt) [20], polo like kinázy I (PlkI) [21], tyrozinkinázy SYK [22] a další.
Přestože se klinický význam celé řady kináz vzájemně překrývá a doplňuje, obecně se podílí na transformaci buněk, iniciaci nádoru, přežití a proliferaci. Zajímavostí je, že i když jsou různé tyrozinkinázy definovány rozdílnými sekvencemi aminokyselin, jejich 3D struktura je relativně konzistentní, což může být příčinou schopnosti řady TKI ovlivnit funkci vícero kináz [9,23,24].
Druhy inhibitorů kinázy
Inhibitory kinázy prokázaly dobrou účinnost v léčbě nádorového onemocnění, zvláště pak ve spojení se specifickými mutacemi. Jejich klasifikace obvykle vychází ze schopnosti katalyzovat přenos fosfátové skupiny z ATP na jednotlivé substráty, které obsahují serinový, threoninový nebo tyrozinový zbytek. Většina z inhibitorů kináz cílí na vazebné místo pro ATP [25,26], některé pak na alosterická místa kináz nebo substrátu [27]. Z klinického hodnocení inhibitorů kináz vyplynulo, že terapeutická odpověď se u jednotlivých pacientů významně liší, což je podmíněno celou řadou vnitřních a vnějších faktorů. Výsledkem je výrazná variabilita intracelulární koncentrace TKI mezi pacienty po podání identické dávky [28−34]. Mezi ovlivňující faktory patří (epi)genetika, pohlaví, etnicita, potrava, současně užívaná léčiva a potravní doplňky, kouření a další. Dle místa působení a způsobu vazby jsou TKI děleny do pěti skupin.
Inhibitory typu I
Účinek inhibitorů kináz typu I vychází z kompetice. Svojí strukturou napodobují purinový kruh adeninového zbytku ATP, interagují tak s aktivním katalytickým místem kináz. Vzniklou vazbou mění strukturní konformaci kinázy, ta brání přenosu fosfátové skupiny z ATP [35,36]. Inhibitory kináz tohoto typu vykazují nízkou selektivitu, čímž se zvyšuje potenciál rozvoje nežádoucích účinků (NÚ), např. kardiotoxicity [37,38]. Do této skupiny patří bosutinib, krizotinib, dasatinib, erlotinib, gefitinib, lapatinib, pazopanib, ruxolitinib, sunitinib a vemurafenib.
Inhibitory typu II
Účinek této skupiny kináz je zprostředkován interakcí s katalytickým místem neaktivované kinázy, a to v jiném místě, než kde probíhá fosforylace [39]. Tyto inhibitory interagují reverzibilně s cílovou kinázou, což vede k tvorbě jednoduchých a/nebo vícenásobných vodíkových vazeb [35,39]. Vazba vykazuje vysoký stupeň selektivity, proto i nižší vyjádření NÚ. Do této skupiny patří imatinib a sorafenib.
Inhibitory typu III − alosterické inhibitory
Třetí skupina inhibitorů kináz se váže na specifická vazebná místa mimo katalytickou oblast pro ATP [40]. Skupina vykazuje vysoký stupeň selektivity, dobrou snášenlivost a nižší míru rezistence na farmakoterapeutickou odpověď [41]. Kompetice s ATP nemůže v tomto případě zabránit jejich interakci s cílovou kinázou. Do této skupiny patří např. trametinib.
Inhibitory typu IV − inhibitory řízené substrátem
Tyto inhibitory se váží opět mimo vazebnou oblast pro ATP, a to v místě substrátu dané kinázy. Tyto inhibitory proto nesoutěží s ATP a nabízejí opět vyšší stupeň selektivity [42], např. ONO12380.
Inhibitory typu V − kovalentní inhibitory
Inhibitory kinázy vytvářejí ireverzibilní kovalentní vazbu s aktivním místem, kdy vzniká nevratný komplex enzym–inhibitor [43−45]. Do této skupiny spadá afatinib, ibrutinib a nová molekula neratinib.
Vlastnosti inhibitorů kináz
Mezi společné vlastnosti TKI patří perorální podání a dlouhý biologický poločas, který umožňuje podání obvykle 1−2× denně. Biologická dostupnost je nicméně poměrně variabilní a pohybuje se zpravidla v rozmezí 70−80 %. Všechny TKI jsou charakterizovány velkými rozdíly v ploše pod křivkou plazmatické koncentrace (area under the curve, AUC), což ústí ve variabilitu v celkové expozici i s následným ovlivněním účinnosti a toxicity [46]. Systémová expozice je ovlivněna následujícími faktory:
- intestinálně absorbovanou frakcí (význam potravy, membránových transportérů, léčiv),
- vazbou na proteiny (albumin, alfa 1 kyselý glykoprotein − AGP),
- up take do hepatocytu (membránové transportní systémy),
- jaterním metabolismem (efekt prvního průchodu játry, CYP450),
- biliární exkrecí.
Jak terapeutická odpověď, tak také výskyt a závažnost NÚ korelují s obvyklými farmakokinetickými ukazateli (AUC, maximální plazmatická koncentrace [cmax], ustálená plazmatická koncentrace apod.). Proto dle předpokladu řada studií prokázala přínos terapeutického monitorování léčiv (therapeutic drug monitoring, TDM) u TKI. Dávkování vycházející ze znalosti plazmatických koncentrací léčiva prokázalo jak vyšší účinnost, tak současně nižší výskyt a závažnost NÚ [32−34,47−51]. Nicméně je třeba pamatovat na možnou diskrepanci, která je dána skutečností, že stanovená plazmatická koncentrace TKI nemusí vždy přesně korelovat s tkáňovou/buněčnou koncentrací TKI. Ačkoliv se nesporně jako výhodnější jeví stanovení intracelulární koncentrace TKI, proti stojí dosud omezené vyšetřovací možnosti [49]. Navzdory těmto uvedeným faktorům jsou TKI podávány ve standardních iniciálních dávkách.
Klíčová role membránových transportérů?
Jak bylo uvedeno výše, biologická dostupnost je u většiny TKI variabilní. Absorpce je ovlivněna řadou faktorů, mezi které patří především současný příjem potravy a aktivita influxních/efluxních membránových přenašečů. Ty jsou nezbytné nejen pro absorpci z trávicího traktu, ale rovněž pro vstup TKI do cílových buněk a pro eliminaci z nich. Průnik TKI do cílové buňky je tedy základním předpokladem pro vazbu TKI s ATP vazebným místem tyrozinkináz.
Lidský genom kóduje více než 400 takových transportérů. U 20 z nich je prokázána spojitost s transportem léčiv přes membránu [52]. Transportní systémy se nacházejí na membráně enterocytů, hepatocytů, proximálních tubulů renálních buněk, endotelu, hematoencefalické bariéry. Svoji klíčovou roli sehrávají nejen ve vztahu k účinnosti (průnik TKI přes membránu do cílové buňky a následná inhibice kinázy), ale také k toxicitě a rezistenci.
Ve vztahu k TKI mají dominantní postavení především tzv. ABC pumpy (ATP binding cassette) a SLC přenašeče (Solute Carrier). V případě ABC přenašečů se jedná o ATP dependentní transportní systém, který plní efluxní funkci (např. na povrchu enterocytů brání absorpci). Často bývá označován jako primární aktivní transport. Kromě vztahu k účinnosti je dáván do souvislosti také s mnohočetnou rezistencí na léky (multidrug resistance) u nádorových buněk. Do skupiny ABC pump je řazen P glykoprotein (kódovaný ABCB1/MDR1) a BCRP (breast cancer resistance protein – kódovaný ABCG). Většina TKI (kromě kabozantinibu, ibrutinibu, regorafenibu, ruxolitinibu, trametinibu a vandetanibu) jsou substráty těchto systémů [52,53]. Nicméně u řady léčiv, u kterých byl sice v prostředí in vitro popsán vliv P glykoproteinu (především pak u bosutinibu, nilotinibu a sorafenibu), je dnes skutečný význam in vivo zkoumán [52]. Přenašeče ABCB1 a ABCG2 jsou spojovány také s rezistencí k cytostatikům [54−56]. I proto FDA doporučuje význam těchto dvou systémů v průběhu klinického zkoušení nových léčiv sledovat [57].
Druhou velkou skupinou jsou SLC přenašeče. Zdrojem energie těchto systémů je přesun iontů koncentračním a elektrochemickým spádem, tzv. sekundární aktivní transport. Význam systémů spočívá ve zprostředkování vstupu (uptake) látek do buněk (enterocytu, hepatocytu). Do této skupiny patří následující systémy:
- OATP 1B1 (SLC01B1), OATP 1B3 (SLC01B3) – organic anion transporting polypeptides,
- OCT1 (SLC22A1) – organic cation transporters.
Ačkoliv se zdá, že většina TKI nemůže být považována za významné substráty SLC přenašečů, ve vztahu k farmakokinetice je přesto potřeba s jejich případným vlivem počítat především v případě axitinibu (OATP1B1, OATP1B3), regorafenibu (OATP1B1), nintedanibu a dasatinibu (OCT1) [52].
Do popředí se v posledních letech stále častěji dostává možnost průniku TKI membránou prostřednictvím pasivní difuze. Tomu by nasvědčovala i skutečnost, že řada TKI není při fyziologickém pH 7,4 polarizována [58−61]. Již dnes je průnik difuzí potvrzen například u palbociklibu [62]. Ve vztahu k nádorovým onemocněním není dosud ani přesně vyjádřen vliv onemocnění samotného na expresi genů kódujících proteinové přenašeče. Zastoupení i aktivita přenašečů mohou být totiž odlišné u zdravých, nádorových buněk a metastáz a mohou mít souvislost s nižší odpovědí u onkologických pacientů na podaná léčiva v důsledku nedostatečné intracelulární koncentrace TKI (snížený influx a zvýšený eflux). I v tomto případě by měly být tyto odlišnosti podrobeny dalšímu zkoumání. Stejně jako otázky, zda deficit jednoho transportního systému nemůže být kompenzován jiným či zda je kapacita transportních systémů dostatečná [52].
Distribuce, metabolismus, eliminace
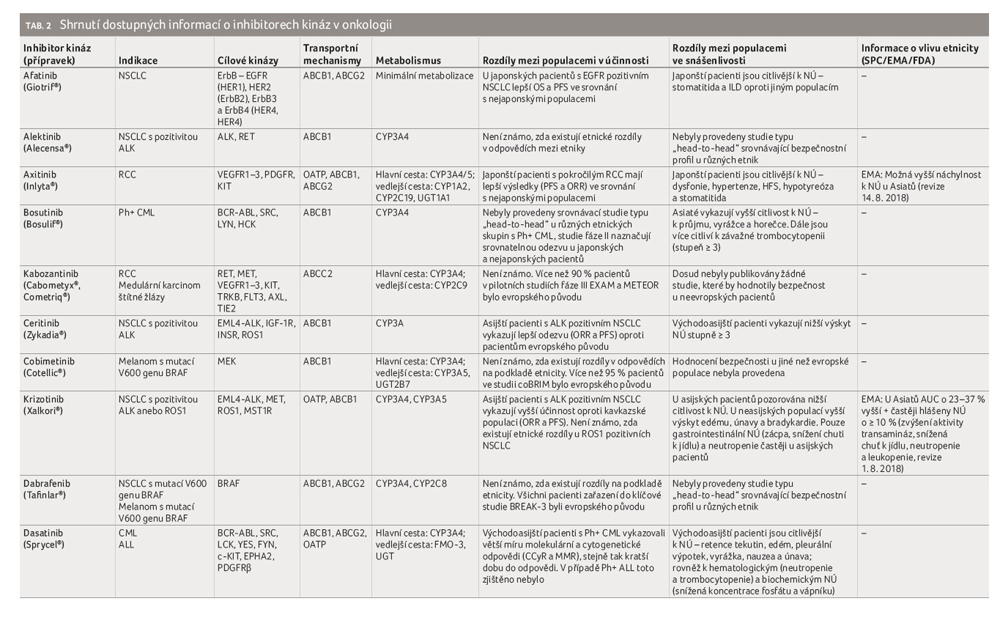
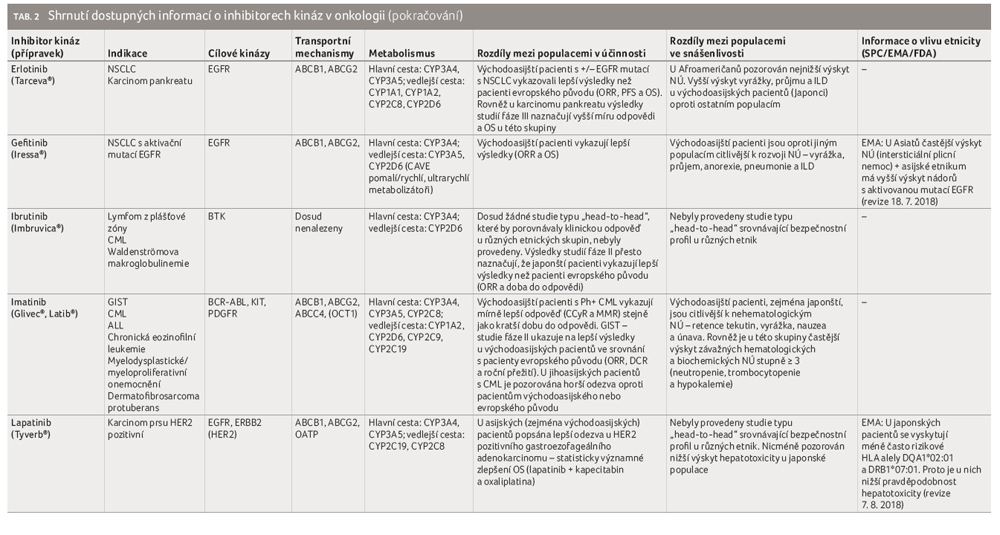
Pro většinu TKI platí, že zdánlivý distribuční objem dosahuje vysokých hodnot, obvykle v rozmezí 100−1 000 litrů. Jak bylo uvedeno, biologický poločas je u většiny TKI relativně dlouhý (24−48 hodin). Mezi léčiva s kratším biologickým poločasem patří např. axitinib, dabrafenib, dasatinib, ibrutinib, nintedanib, ruxolitinib. Naopak mezi léčiva s výrazně delším biologickým poločasem se řadí trametinib a vandetanib (4 dny, resp. 10 dní) [52,63]. Inhibitory tyrozinkináz jsou z více než 90 % vázány na plazmatické bílkoviny. Hlavní roli zde hraje albumin a AGP. Patologické stavy (infekce, nádor) mohou vést k vyšší plazmatické koncentraci AGP, následně tedy mohou ovlivnit účinnost a toxicitu. Na metabolismu TKI se podílejí v naprosté většině v první fázi enzymy cytochromu P450. Dominantní postavení má izoforma CYP3A4/5. Význam dalších izoforem – CYP1A1, CYP1A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6 – je menší, viz tabulku 2 (pod článkem). Ve druhé fázi pak probíhá metabolizace prostřednictvím UGP (uridin difosfát glukuronosyltransferáza). Pro léčebnou odpověď a toxicitu má nezanedbatelný význam také to, zda vznikají metabolity neaktivní, či metabolity s vyšší či nižší aktivitou. Příkladem mohou být sunitinib, imatinib, sorafenib a erlotinib, ze kterých vznikají metabolity s aktivitou podobnou původní molekule [64,65]. Výsledná klinická odpověď se tedy rovná součtu aktivit TKI a vzniklého metabolitu. Inhibitory tyrozinkináz jsou primárně eliminovány jaterním metabolismem a biliární exkrecí zprostředkovanou P glykoproteinem, méně než 10 % je vyloučeno močí v nezměněné formě.
Rozdíly ve farmakokinetice na podkladě etnicity – vnitřní faktory
S hlubším poznáním a širší
zkušeností s používáním TKI u různých etnik roste
i význam etnicity ve vztahu k variabilitě
terapeutických odpovědí [66−69]. I proto je dnes etnicita
řazena mezi jeden z vnitřních faktorů predikujících
účinnost a toxicitu TKI. Hlavní pozornost výzkumu
zahrnujícího oblast farmakokinetiky je dnes soustřeďována
na rozdíly v expresi a/nebo v aktivitě genů, které
kódují enzymy ovlivňující metabolismus TKI (např. cytochrom
P450), a membránových transportních proteinů. Ve vztahu
k rozdílům ve farmakodynamických parametrech dominují
geny kódující proteiny, které se podílejí na samotném
účinku – často se jedná o cílové struktury působení
TKI. Mezi již dobře zdokumentované rozdíly patří například
odlišná prevalence aktivačních mutací genu EGFR u pacientů
s nemalobuněčným karcinomem plic (NSCLC). Pro kavkazskou
populaci se udává výskyt těchto mutací přibližně u 10−20 %
pacientů [70,71]. Méně známým faktem je, že se tyto „klasické“
aktivační mutace genu (deleční v exonu 19 a L858R
v exonu 21) vyskytují častěji u Asiatů, respektive
u východoasijských pacientů (30−40 %) [71–73]. Uvedené
aktivační mutace významným způsobem předurčují dobrý efekt
léčby, a mohou tedy alespoň zčásti vysvětlit pozorovanou
lepší odpověď na erlotinib a gefitinib u této
populace [74−81]. Rozdíly ve výskytu dalších mutací genů
u pacientů s NSCLC u různých etnik uvádí tabulka 3 [71].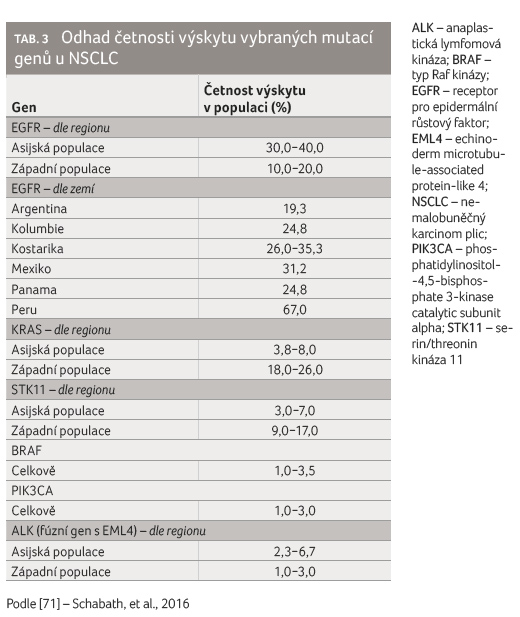
Rozdíly v absorpci
Zásadní význam zde hraje především systém membránových přenašečů BCRP a P glykoproteinu. Aktivita těchto systémů může být různá u různých etnik, což může být částečně vysvětleno rozdílným zastoupením variantních alel jednotlivých polymorfismů genů kódujících zmíněné proteiny. Membránový přenašeč BCRP je kódován genem ABCG2. Mezi jeho substráty patří řada TKI − afatinib, axitinib, dabrafenib, dasatinib, erlotinib, gefitinib, imatinib, lapatinib, nilotinib, osimertinib, pazopanib, ponapanib, sorafenib a sunitinib. Mezi významné polymorfismy genu ABCG2 patří například jednonukleotidové polymorfismy (single nucleotide polymorphism, SNP) ABCG2 34G>A (Val 12 Met, rs 2231137) a 421 C>A (Gln 141 Lys, rs2231142). V obou případech jsou variantní alely A spojeny se sníženou expresí a s efluxní aktivitou BCRP. To může vést ke zvýšení plazmatické a následně i buněčné koncentrace BCRP uvedených substrátů v cílových buňkách. Variantní alely uvedených polymorfismů se vyskytují u východoasijské populace s výrazně vyšší četností. Výskyt variantní alely A u polymorfismu ABCG2 34 G>A byl zjištěn pouze asi u 3 % kavkazské populace, naopak ve východoasijské populaci nalezneme tuto alelu až u poloviny pacientů. Možná i proto byla u této skupiny pacientů potvrzena lepší terapeutická odpověď na léčbu imatinibem [82,83] či vyšší toxicita u gefitinibu [84]. I v případě SNP ABCG2 421 C>A bylo vyšší zastoupení pacientů s genotypem AA častější u východoasijské populace (Japonci 34,1 %, Číňané 29,2 %) oproti populaci kavkazské (7−11,1 %). To je spojováno s vyšší systémovou expozicí imatinibu a sunitinibu [85−87]. To opět dle řady studií může souviset s lepší terapeutickou odpovědí na imatinib či s nižší snášenlivostí sunitinibu, erlotinibu a gefitinibu u východoasijských pacientů [83,88−93].
Východoasijská populace
Tato populace má významné postavení ve farmakogenetických studiích s TKI, neboť právě naprostá většina rozdílů v účinnosti a toxicitě byla dosud potvrzena mezi kavkazskou a východoasijskou populací. Etnicky do této populace patří osoby původem z Číny, Japonska, Koreje, ale rovněž z Mongolska a Tchaj wanu. U jiných populací je zkušenost s podáváním TKI, především pro vysokou cenu, velmi omezená. Východoasijská populace celosvětově tvoří až pětinu populace a asi 40 % všech Asiatů. V rámci této skupiny pak dominuje čínské etnikum Han (1,3 miliardy lidí = 95 % Číňanů). Mimo původní region můžeme nalézt tuto populaci např. v USA (3,8 mil.), Kanadě (1,5 mil.), Vietnamu (0,8 mil.), Rusku (1 mil.), Francii (0,7 mil.). V kontextu České republiky pak hovoříme o počtu přibližně 20 tisíc osob, kdy převládají občané Mongolska a Číny [94].
Interetnické rozdíly ovlivňující distribuci TKI
Jedním z prokázaných rozdílů ve farmakokinetických parametrech na podkladě etnicity, které mohou ovlivňovat distribuci TKI v organismu, je rozdílná koncentrace AGP. U východoasijské populace byla zjištěna nižší koncentrace AGP v krvi oproti populaci kavkazské [95]. To vede například v případě imatinibu ke zvýšení volné (nevázané) frakce [96−98]. V důsledku toho dochází k rychlé redistribuci do tkání, ke zvýšení účinnosti imatinibu v léčbě CML, ale také k vyšší toxicitě u východoasijských pacientů [98]. Mezi další faktory, které mohou mít vliv na distribuci TKI a u kterých byly potvrzeny rozdíly mezi etniky, patří například index tělesné hmotnosti (BMI), hmotnost a množství/procento tělesného tuku. Uvedené parametry souvisejí se změnami v distribučním objemu a v eliminaci. Na základě studií bylo u východo a jihoasijských pacientů zjištěno, že i při nižší hodnotě BMI a LBM (lean body mass, hodnota získaná odečtením množství tělesného tuku od celkové tělesné hmotnosti) mají vyšší podíl tělesného tuku [99−102]. U TKI lipofilního charakteru se tedy zvětšuje zdánlivý distribuční objem. To bývá obvykle dáváno do souvislosti s vyšší četností výskytu NÚ, případně s jejich závažností u Asiatů [103−110].
Rozdíly na podkladě etnicity – vnější faktory
S onkologickou léčbou je spojována, jako snad s žádnou jinou léčbou, alternativní, komplementární a v posledních letech také integrativní medicína. V posledním případě se sice jedná o kombinaci alternativní a komplementární medicíny, nicméně je podložena daty o efektivitě a také je uplatňována v souladu se standardní terapií a se souhlasem lékaře. Využívány jsou kromě řady fytofarmak také další nefarmakologické metody, mezi něž patří například akupunktura, akupresura, jóga a léčebný program snižování stresu (mindfulness based stress reduction, MBSR). Z řady studií vyplývá, že během onkologické léčby užívá přípravky z komplementární a alternativní medicíny 70−90 % pacientů. Jako zcela zásadní se jeví fakt, že téměř tři čtvrtiny pacientů o této skutečnosti svého onkologa neinformují [111,112]. To může být problematické s ohledem na zjištění americké studie, že až čtvrtina pacientů sahajících v průběhu chemoterapie k alternativní léčbě užívá stran interakcí potenciálně nebezpečné přípravky [113]. Většina TKI je metabolizována cestou cytochromu P450 (nejčastěji CYP3A4) a současně se jedná o substráty řady transportních přenašečů (P glykoprotein apod.). Jejich interakční potenciál je tedy vysoký. Klasickým příkladem rizikového fytofarmaka je třezalka tečkovaná. Ta je silným induktorem jak CYP3A4, tak CYP2C9, což ovlivňuje i celou řadu TKI − např. imatinib, sunitinib, pazopanib, gefitinib, erlotinib aj. [114].
Existují však etnicky podmíněné
rozdíly v přístupu k alternativní a komplementární
léčbě. U asijské populace byla prokázána obecně vyšší
míra adherence k doplňkům stravy a k fytofarmakům
oproti ostatním populacím. Mezi tradičně využívané patří
zelený čaj, ženšen a sójové produkty [115,116]. Jak je
znázorněno v grafu 1,
který spojuje závěry více studií, jedná se o významné
rozdíly, s nimiž je třeba při léčbě TKI počítat
[117−120].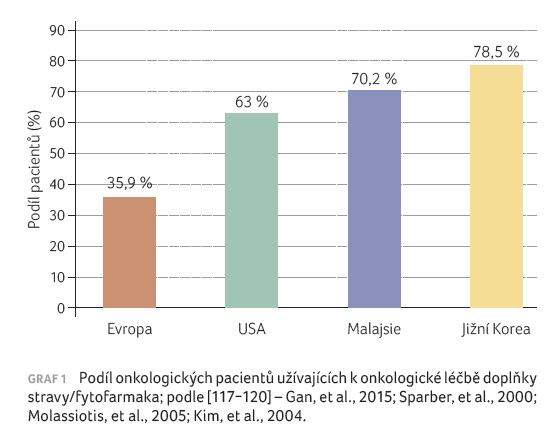
Dalším významným faktorem, který se podílí na terapeutické odpovědi některých TKI, je kouření. I zde existují mezi etniky velké rozdíly. Ještě větší rozdíly jsou pak při srovnání mezi etniky s ohledem na pohlaví. Například v ČR kouří okolo 24 % populace (27,3 % mužů vs. 21,1 % žen), zatímco v Číně v roce 2010 dle statistik kouřilo 52,9 % mužů, ale jen 2,4 % žen [121,122]. Polycyklické aromatické uhlovodíky v tabákovém kouři mají potenciál ovlivnit metabolismus řady látek, především pak prostřednictvím indukce CYP1A1 a CYP1A2 [123]. Tato skutečnost je v přímé souvislosti s metabolismem erlotinibu, kdy byla u kuřáků prokázána vyšší clearance. Výsledkem byla 2,8krát nižší hodnota AUC a 8,3krát nižší hodnota průměrné plazmatické koncentrace oproti nekuřákům [124,125]. Metaanalýzou bylo dle očekávání potvrzeno, že kouření představuje rizikový faktor snižující účinnost léčby jak erlotinibu, tak gefitinibu (zkrácení přežití bez progrese nemoci – progression free survival, PFS) [126].
Následující text se pokusí sumarizovat závěry řady studií, které zkoumaly variabilitu v terapeutických odpovědích inhibitorů kináz u různých etnik, a upozornit na významné rozdíly u vybraných skupin nádorových onemocnění (CML, NSCLC, RCC).
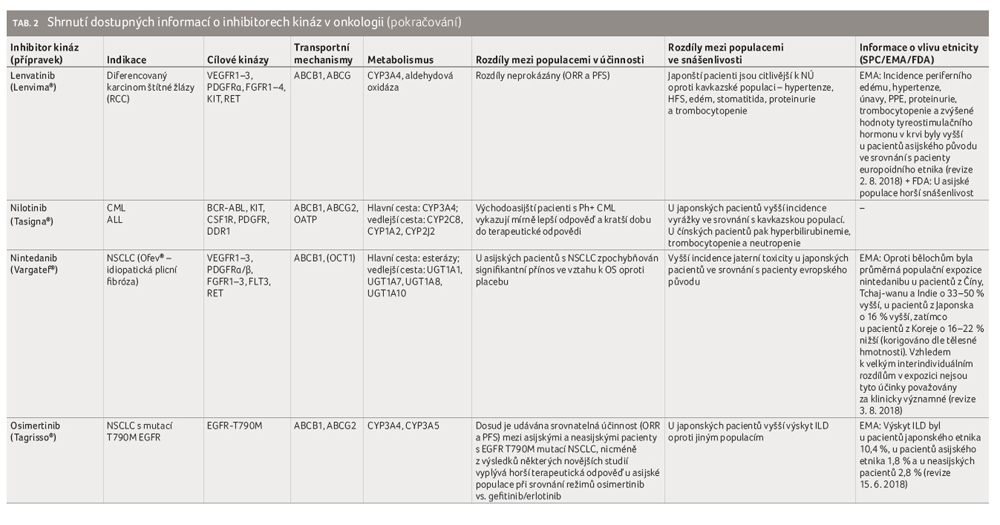
Etnicitou podmíněná variabilita v odpovědích na cílenou terapii CML
Při podrobnějším rozboru pacientů s CML byly zjištěny určité rozdíly mezi populacemi. Americká studie například poukázala na celkově nižší výskyt CML u asijské populace oproti populaci kavkazské (0,4−0,9 případu vs. 1,5 případu na 100 000 obyvatel). Zajímavým zjištěním pak bylo, že průměrný věk při stanovení diagnózy byl u Asiatů výrazně nižší (36−55 let u Asiatů vs. 65 let u kavkazské populace) [103].
Ačkoliv se v případě imatinibu
jedná o první zaregistrovaný TKI (1. generace TKI), jeho
role v terapii CML je stále významná. Charakteristickým
znakem nádorových buněk u CML je až 95% pravděpodobnost
přítomnosti filadelfského chromozomu (Ph+, Ph1) [127]. Ten vzniká
translokací části 9. chromozomu nesoucí protoonkogen c abl
na 22. chromozom, kde je umístěn gen bcr. Výsledkem je
fúzní gen bcr abl, který kóduje tyrozinkinázu stejného
označení. Inhibice aktivity tyrozinkinázy BCR ABL vede
k zastavení množení a k apoptóze leukemických
buněk. Imatinib nicméně blokuje aktivitu i dalších kináz,
viz tabulku 3. Odlišné
odpovědi na imatinib mezi jednotlivými populacemi jsou
zdokumentovány jak z hlediska farmakodynamického, tak
farmakokinetického. Stran farmakodynamických odlišností byla
popsána lepší terapeutická odezva u východoasijské
populace oproti populaci evropské [103,128]. Jako určitý paradox
se může jevit naopak horší odezva u jihoasijské populace.
Vysvětlením může být rozdílné zastoupení variantní alely T
genu BCL2L11/BIM (465C>T), který kóduje regulační protein
procesu buněčné apoptózy Bcl 2 like protein 1.
Variantní alela T je spojována s horší terapeutickou
odpovědí a její zastoupení je nejvyšší právě
u jihoasijské populace (56,9 %), následuje populace evropská
(29,6–40,7 %) a východoasijská (Čína 12,8−14,6 %,
Japonsko 8,4 %) [82]. Horší odpověď u jihoasijské
populace může rovněž souviset s variabilitou exprese genu
pro interferon gama (IFNγ), a to prostřednictvím vlivu
na proliferaci hematopoetických kmenových buněk. V konečném
důsledku dochází ke změně citlivosti CML k imatinibu.
Genotyp CC polymorfismu IFNG 1616C>T (rs2069705) je
dáván do vztahu s lepší terapeutickou odpovědí
(v parametrech kompletní cytogenetické odpovědi CCyR a tzv.
velké molekulární odpovědi MMR) [129]. Zmíněný genotyp se
častěji vyskytuje u východoasijské populace (Japonsko 78 %
a Čína 53–66 %) oproti Evropanům (10 %) a populaci
jihoasijské (6 %) [82].
Ve vztahu k toxicitě byla popsána vyšší četnost nehematologických NÚ (retence tekutin, vyrážka, nevolnost, únava) u Asiatů, zvláště Japonců. Asijská populace byla obecně rovněž citlivější k hematologickým NÚ (neutropenie, trombocytopenie a hypokalemie) stupně závažnosti ≥ 3 [103].
Rozdíly ve farmakokinetice imatinibu mezi etniky se promítají do výsledné systémové expozice. Dle očekávání je vyšší systémová expozice spojena jak s lepší terapeutickou odpovědí u pacientů s CML, tak s toxicitou [85−87,130,131]. Rozhodující měrou ovlivňují systémovou expozici transportní systémy (P glykoprotein a BCRP). Jak bylo uvedeno výše, existují mezietnické rozdíly v aktivitě a expresi genů kódujících tyto systémy. Klinicky nejvýznamnější variabilitu podmiňují SNP ABCG2 421C>A (rs2231142) a ABCG2 34G>A (rs2231137). V obou případech je variantní alela A spojována se sníženou expresí a aktivitou membránového efluxního proteinu BCRP, a tudíž i s vyšší systémovou expozicí [83,88]. U kavkazské populace se A alela ABCG2 34G>A vyskytuje pouze výjimečně (asi u 3 %), ve východoasijské populaci se vyskytuje až u poloviny pacientů s CML [82].
Podobně má i AA genotyp ABCG2 421C>A četnější zastoupení ve východoasijské populaci (okolo 30 %) oproti populaci kavkazské (okolo 10 %) [82]. Východoasijští pacienti tedy při podání stejné dávky imatinibu dosahují častěji vyšších plazmatických koncentrací, a tedy i terapeutické odezvy. Význam polymorfismů genu pro P glykoprotein není tak jednoznačný [132]. Pozornost je soustředěna především na SNP ABCB1 2677T>G/A (rs2032582), 3435C>T (rs1045642) a 1236T>C (rs1128503), respektive na haplotyp uvedených tří SNP. V roce 2015 byla publikována data metaanalýzy, která vycházela z údajů od 1 826 pacientů a zkoumala vliv uvedených polymorfismů ve vztahu k účinnosti imatinibu v léčbě CML. Výsledky ukázaly, že alela 2677G nebo alela 3435T předpovídaly horší odezvu na imatinib u pacientů s CML (častěji u jihoasijské a kavkazské populace), zatímco genotyp 1236CC byl spojen s lepší odpovědí u pacientů s CML z asijské oblasti [133]. Tyto nové znalosti se dosud nestaly součástí souhrnů údajů o přípravku (SPC) léčivých přípravků s imatinibem registrovaných FDA i EMA (Evropská léková agentura), nicméně v klinické praxi je dobré s uvedenými rozdíly počítat.
Pro pacienty netolerující imatinib nebo rezistentní na léčbu tímto přípravkem se volí mezi dalšími TKI 2. generace, mezi něž jsou řazeny nilotinib, dasatinib a bosutinib. Rovněž u těchto molekul byly popsány rozdíly mezi etniky jak v účinnosti, tak v toxicitě. Ve studiích in vitro dasatinib až 325× převyšuje účinnost imatinibu a podobně jako imatinib se zdá být účinnější u východoasijské populace [103,134]. Rovněž tak se zdá, že tato populace je citlivější k rozvoji hematologických, nehematologických NÚ (retence tekutin, edémy, pleurální výpotek, vyrážka, únava) a abnormalit biochemických parametrů [103]. Nilotinib se váže s 30× vyšší afinitou ke kinázové doméně než imatinib. Ve studiích in vitro nilotinib inhibuje 32 z 33 klinicky relevantních mutací, které jsou příčinou rezistence na imatinib. Východoasijští pacienti dosahovali mírně lepších výsledků. U japonské populace byla zaznamenána vyšší incidence vyrážky, u čínské populace častěji hyperbilirubinemie, trombocytopenie a neutropenie [135−137].
Ponatinib je multipotentní TKI 3. generace. Účinkuje in vitro u všech mutací rezistentních na imatinib, včetně polyrezistentní mutace T315I. Výsledky studie fáze II naznačují vyšší míru odpovědi u japonské populace a rovněž vyšší incidenci NÚ [138,139].
Bosutinib podobně jako dasatinib je duálním inhibitorem kináz Abl a Src. Studie naznačují srovnatelnou odpověď u pacientů různých etnických skupin [140,141], zatímco citlivost k rozvoji nehematologických NÚ (zejména průjem, vyrážka, horečka) byla vyšší u Asiatů [142].
Etnicitou podmíněná variabilita v odpovědích na cílenou terapii NSCLC
Cílená léčba NSCLC směřuje k ovlivnění mutace signalizační dráhy pomocí receptoru pro epidermální růstový faktor (epidermal growth factor receptor, EGFR) a translokace v genech ALK (anaplastická lymfomová kináza) a ROS1. Celosvětově se uvádí výskyt aktivačních mutací EGFR u asi 20 % pacientů, viz tabulku 3 [143]. Vyšší četnost nicméně najdeme u asijské populace, dále u žen, nekuřáků a u pacientů s adenokarcinomem [144].
Aktivační mutace jsou zodpovědné za nekontrolovaný nádorový růst a mají tendenci vytvářet metastázy v důsledku zvýšené buněčné proliferace a inhibice apoptózy. V současné době jsou uznány jako prediktory účinnosti inhibitorů EGFR. Průkaz těchto mutací je proto nezbytným předpokladem pro indikaci cílené terapie TKI. Ty zabraňují aktivaci receptorů a přenosu signálu na další členy signalizační kaskády. Z detekovaných mutací je nejběžnější delece v exonu 19 a bodová mutace L858R v exonu 21 [145].
Inhibitory EGFR
Gefitinib a erlotinib, 1. generace inhibitorů EGFR, blokují EGFR reverzibilně. Představují ukázkový příklad variability terapeutické odpovědi na podkladě etnicity, neboť v případě východoasijských pacientů byla popsána lepší klinická odpověď ve srovnání s pacienty evropského původu [78,80]. Tato populace byla rovněž citlivější k rozvoji NÚ (kožní projevy, průjem, anorexie, pneumonie). V případě intersticiálních plicních procesů (intersticial lung disease, ILD) byl pak největší výskyt zjištěn u pacientů z Japonska [75,76,146]. Zvýšená míra rozvoje NÚ je dávána do souvislosti s variabilitou genů ABCG2. Jako významné z pohledu etnicity se jeví polymorfismy 34G>A (rs2231137) a 421C>A (rs2231142). Variantní alela A v obou případech odpovídá za nižší expresi a aktivitu efluxního proteinu BCRP a tím za vyšší plazmatické koncentrace uvedených TKI. Genotyp GG prvního z nich převažuje u kavkazské populace (97 %). Naopak ve východoasijské populaci se vyskytuje pouze u poloviny pacientů (42−66 %) [82,84]. Podobně je tomu v případě polymorfismu 421C>A, kdy je genotyp AA častěji detekován u východoasijské populace (29−34 %) ve srovnání s Evropany a s jihoasijskými pacienty (oba asi 10 %) či s Afroameričany (1 %) [93,147].
Erlotinib rovněž prokázal lepší výsledky u východoasijské populace v léčbě NSCLC (i karcinomu pankreatu) ve srovnání s ostatními populacemi. I zde platí, že aktivační mutace (L778P a I821T v exonu 20, K728R a W731X v exonu 19) jsou častější u pacientů z východní Asie (Číňané) než u Evropanů [148]. Výjimečně jsou známa bližší data i pro jinou než asijskou a kavkazskou populaci, neboť byla u erlotinibu pozorována nižší systémová expozice a menší incidence NÚ u Afroameričanů ve srovnání s jinými populacemi [149]. K vysvětlení tohoto rozdílu může přispět jak již zmíněný malý výskyt genotypu AA 421C>A ABCG2, tak rovněž variabilita v genu ABCB1 pro P glykoprotein. Výskyt TTT haplotypu ABCB1 (3435T>C/1236T>C/2677T>G/A) snižuje jeho expresi, výsledkem je snížená clearance, respektive zvýšení systémové expozice. Zastoupení TTT haplotypu se udává do 8,5 % u afroamerické populace v kontrastu s 35−56% zastoupením u jiných populací [150−152]. Rozdíly byly identifikovány i na úrovni biotransformace, jak je zřejmé z práce hledající spojitost s polymorfismem CYP1A1 a CYP3A5. U japonských pacientů, nositelů genotypů CYP1A1 2455GG a/nebo CYP3A5 6986GA/GG, byly zaznamenány významně vyšší plazmatické koncentrace erlotinibu [153]. Tyto varianty jsou četnější u východních Asiatů ve srovnání s evropskou a afroamerickou populací [82].
Afatinib je řazen do 2. generace inhibitorů EGFR. Jedná se o vysoce selektivní ireverzibilní blokátor ErbB rodiny (receptory ErbB1 = EGFR, ErbB2 = HER2, ErbB4). Má prokázanou účinnost jak u běžných mutací EGFR, tak i u mutací rezistentních (T790M). Při interetnickém srovnání měli z afatinibu největší prospěch pacienti původu japonského ve srovnání s ostatními populacemi, současně se u nich ale ve větší míře projevovaly NÚ, jako je stomatitida či ILD [154,155].
V krátké době bude na trh uveden další TKI 2. generace – dacomitinib. Stejně jako v případě afatinibu by se mělo jednat o ireverzibilní inhibitor celé rodiny EGFR. Údaje vycházejí z výsledků studie ARCHER 1050 srovnávající dacomitinib (n = 227) s gefitinibem (n = 225) u východoasijské populace, které prokázaly klinicky významné zlepšení průměrného PFS (14,7 měsíce vs. 9,2 měsíce), nicméně přímé srovnání s jinou populací chybí [156].
Osimertinib, zástupce 3. generace inhibitorů EGFR, se ireverzibilně váže na EGFR. Účinkuje na senzitivní mutace EGFR včetně rezistentní mutace T790M. Díky nižší citlivosti k divokému typu EGFR můžeme očekávat menší intenzitu NÚ (vyrážka, průjem). Význam etnicity není u osimertinibu dosud zcela znám, dokonce byly publikovány odlišné závěry. Nicméně z výsledků nejnovější studie, která byla zveřejněna začátkem roku 2018, vyplývá horší terapeutická odpověď u asijské populace oproti jiným populacím při srovnání režimů osimertinib vs. gefitinib/erlotinib [157].
Inhibitory ALK
Jiným řídícím onkogenem u pacientů s NSCLC je ALK. Tento onkogen může vytvářet fúzní gen s genem EML4 (echinoderm microtubule associated protein like 4) [158]. U nemocných s NSCLC se objevuje translokace genu ALK ve zhruba 5 % [159]. Tak jako v případě mutací EGFR se i tato mutace častěji vyskytuje u žen mladšího věku, nekuřáků a u nemocných s adenokarcinomem. Z výsledků řady studií rovněž vyplývá různé zastoupení mutací EML4 ALK u různých populací (Asiaté 2,3−6,7 %, kavkazská populace 1,0−3,0 %) [160,161].
Do 1. generace inhibitorů ALK je řazen krizotinib. Dále inhibuje receptor pro růstový faktor pro hepatocyty (HGFR, c Met) RTK, ROS1 (c ros) a Recepteur d’Origine Nantais (RON) RTK. Léčba krizotinibem u ALK pozitivních asijských pacientů byla spojena s lepšími výsledky oproti jiným populacím a současně se ukázali být pacienti této skupiny méně citliví k výskytu NÚ (edémy, bradykardie) [162,163]. Pouze v případě gastrointestinálních NÚ tomu bylo opačně [164].
ALK inhibitory 2. generace – ceritinib a alektinib – účinkují na nádorové buňky s rezistencí vůči krizotinibu. Existují dvě hlavní skupiny příčin rezistence – tzv. ALK dominantní (ALK mutace – L1196M, G1269A, I1171T a S1206Y, ALK amplifikace, metastázy do CNS) a ALK nedominantní (mutace či amplifikace jiných řídících genů, popřípadě přeměna v sarkomatoidní typ nádoru). U ceritinibu byla pozorována lepší terapeutická odpověď u populace asijské a asijští pacienti také méně inklinovali k rozvoji NÚ vyššího stupně závažnosti [163]. Alektinib působí nejen proti ALK, ale i proti dalším kinázám (LTK – leukocyte receptor tyrosine kinase, GAK – cyclin G associated kinase). U alektinibu nebyl význam etnicity potvrzen.
Inhibitory angiokináz
Zajímavých výsledků bylo také dosaženo u nintedanibu, inhibitoru angiokináz (VEGFR 1–3, PDGFR α/ß, FGFR 1–3 a dalších). Ze studie LUME Lung 1 srovnávající léčbu kombinací docetaxelu s nintedanibem vs. podávání docetaxelu a placeba vyplynulo, že asijští pacienti s NSCLC nemusejí mít signifikantní terapeutický přínos ve vztahu k celkovému přežití (overall survival, OS) oproti placebu [165]. Na druhou stranu u nich byla pozorována vyšší incidence hepatotoxicity [166,167]. S ohledem na skutečnost, že se jednalo o rozsáhlou multicentrickou studii, které se zúčastnilo 1 314 pacientů (81 % kavkazská populace, 18 % Asiaté, 1 % ostatní), mají výsledky poměrně silnou váhu.
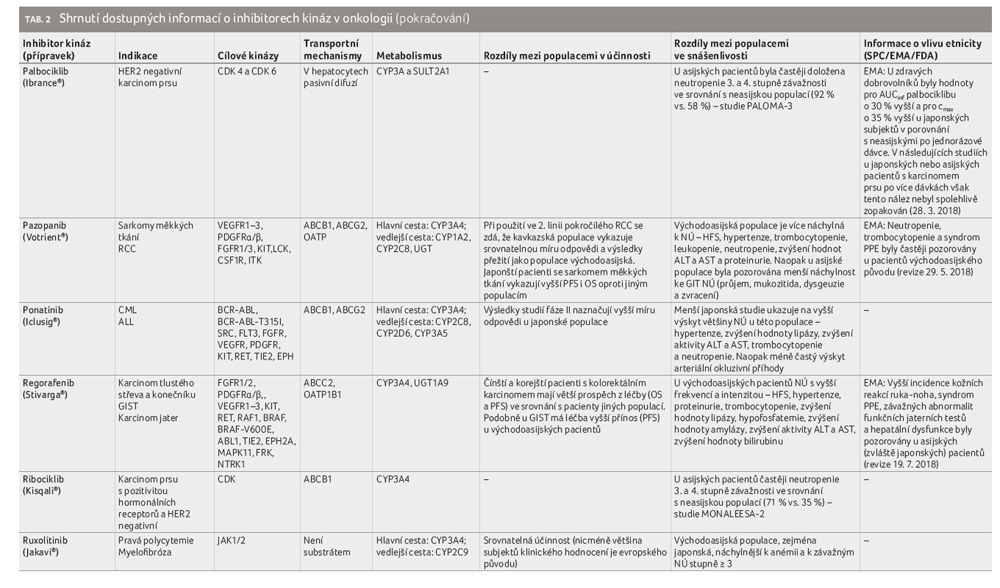
Etnicitou podmíněná variabilita v odpovědích na cílenou terapii renálního karcinomu
Cílená léčba TKI našla uplatnění v terapii lokálně pokročilého inoperabilního a metastatického renálního karcinomu (renal cell carcinoma, RCC). Ačkoliv v této indikaci dostupné TKI cílí především na klíčovou signální dráhu v procesu angiogeneze zprostředkovanou VEGF (vascular endothelial growth factor, vaskulární endoteliální růstový faktor), jedná se o multikinázové inhibitory.
Sunitinib je multikinázový inhibitor (tab. 2). I když nebyl nalezen rozdíl v terapeutické odpovědi mezi pacienty neasijského a východoasijského původu (time to progression, TTP; PFS), u jihoasijských pacientů tomu tak nebylo, neboť u nich byly prokázány horší výsledky léčby (overall response rate, ORR; PFS) oproti uvedeným populacím [107,109,168]. Na druhou stranu léčba asijských pacientů byla obecně spojena s častějším rozvojem NÚ (trombocytopenie, neutropenie, anémie, syndrom ruka noha, hypertenze, průjem, únava, mukozitida), i s jejich závažností [109,169]. Tento rozdíl lze vysvětlit polymorfismem jak v genech kódujících BCRP a P glykoprotein, tak v polymorfismech genů cílových receptorů VEGFR, PDGFR, FLT3 nebo biotransformačních enzymů cytochromu P450 CYP1A1 a 3A5. Ačkoliv SPC nezmiňuje potřebu úpravy dávkování na základě etnicity, českým lékařům je rozdíl v účinnosti a v toxicitě u pacientů asijského původu znám [170]. Dokonce je na základě výsledků studie z roku 2015 doporučeno u asijských pacientů s karcinomem ledvin začínat léčbu redukovanou iniciální dávkou 37,5 mg oproti běžné dávce 50 mg [171]. Ze závěrů studie totiž vyplynulo, že redukce dávky sníží rizika vážných NÚ při srovnatelném účinku.
Sorafenib je multikinázový inhibitor (tab. 2). Přestože se metabolizuje podobně jako sunitinib na farmakologicky aktivní metabolit, nepředpokládá se ovlivnění výsledného účinku. Význam etnicity ve vztahu k účinnosti terapie RCC je dosud poznamenán protichůdnými výsledky. Existují studie popírající etnické rozdíly, nicméně některé práce naznačují lepší odezvu u pacientů asijského původu [172−175]. Z hlediska rozvoje NÚ jsou východoasijští pacienti náchylnější ke vzniku syndromu ruka noha, k průjmu, hypertenzi, alopecii a anorexii [108,110,176]. Dosud byly identifikovány polymorfismy 15 kandidátních genů, které mají souvislost s terapeutickým účinkem (zejména pro VEGF a VEGFR) či s toxicitou sorafenibu (nejvíce pro VEGF a ABCB1) [177]. Přesto existuje řada dalších geneticky podmíněných odlišností mezi etniky, především pak ve vztahu k toxicitě. Například ve studii hodnotící efekt sorafenibu v indikaci hepatocelulárního karcinomu byla zjištěna souvislost variability genu pro glukuronizační enzym UGT1A9 IVS1 37431 s rozvojem syndromu ruka noha. Genotyp AA, který je spojen s rozvojem tohoto NÚ s vyšším stupněm závažnosti, se častěji vyskytuje u východoasijské populace [108].
Pazopanib je multikinázový inhibitor. Byl pozorován srovnatelný klinický účinek nezávislý na etnickém původu, zatímco citlivost na některé NÚ (syndrom ruka noha, hypertenze, trombocytopenie, leukopenie, neutropenie, zvýšení aktivity jaterních enzymů ALT/AST, proteinurie) se typicky ve větší míře vyskytovala u asijské populace na rozdíl od evropské, která byla více zasažena gastrointestinálními NÚ (průjem, mukozitida, zvracení) [106].
Axitinib je selektivní inhibitor 2. generace, který blokuje VEGFR při nízkých koncentracích. V celé řadě publikovaných prací se objevují data poukazující na lepší účinnost u pacientů z Japonska oproti ostatním skupinám. Současně byli i více postiženi NÚ, jako jsou dysfonie, hypertenze, syndrom ruka noha, poruchy funkce štítné žlázy, stomatitida [173−175,178].
Kabozantinib je multikinázový inhibitor s aktivitou namířenou proti MET, AXL a třem receptorům VEGF. Aktivace receptorů MET a AXL se podílí na rezistenci nádorových buněk vůči TKI a je spojena s agresivnějším chováním nádoru. Bylo prokázáno, že ke zvýšené expresi MET a AXL dochází během léčby sunitinibem (což však lze extrapolovat i na ostatní typy anti VEGF terapie) [179]. Není znám vliv na účinnost a toxicitu z hlediska etnicity.
Lenvatinib, multikinázový inhibitor s převahou pro VEGFR2 a VEGFR3, byl dosud registrován pro léčbu metastatického karcinomu štítné žlázy, registrace pro metastatický RCC se očekává. Nelze hodnotit existenci rozdílu v účinnosti mezi populacemi s ohledem na skutečnost, že studií se účastnili převážně pacienti evropského původu [180].
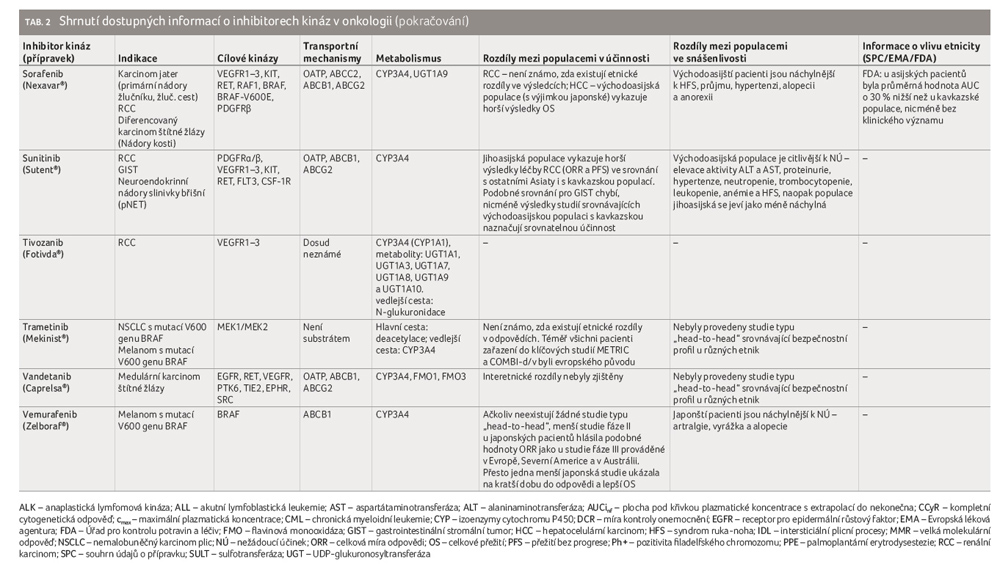
Závěr
Role etnicity ve vztahu k účinnosti a snášenlivosti byla již u řady TKI prokázána. Variabilita je obvykle podmíněna odlišnostmi v genetické výbavě jednotlivých populací. Nejčastěji se jedná o rozdílný výskyt variantních alel polymorfismů genů, které mají souvislost s farmakokinetikou či s farmakodynamikou TKI. Variabilita může být nicméně podmíněna také celou řadou dalších vnitřních i vnějších faktorů (stravovací návyky, užívaná fytofarmaka, koncentrace AGP, poměr tělesného tuku a další). Největší zkušenosti s TKI jsou u kavkazské a (východo)asijské populace, i proto je nejvíce dostupných informací o rozdílné účinnosti a snášenlivosti právě mezi těmito populacemi. Pro další populace data spíše chybějí. U řady registrovaných léčivých přípravků je již zmínka o vlivu etnicity součástí SPC. V případě axitinibu, gefitinibu, krizotinibu, lenvatinibu, osimertinibu, pazopanibu, regorafenibu je uvedena zmínka o vyšší citlivosti a častějším výskytu NÚ u asijské populace. Pouze v případě lapatinibu je popsáno nižší riziko hepatotoxicity ve srovnání s kavkazskou populací. U ostatních zástupců či populací vliv etnicity buď prokázán nebyl, nebo dosud neexistují dostatečně silná data. Snadnou implementaci nových informací rovněž komplikuje nejednotnost uspořádání studií a často malý počet pacientů. Obecně se ale jeví asijská populace jako citlivější k rozvoji NÚ, také je u ní obvykle pozorována lepší terapeutická odpověď (ORR, OS, PFS). Jasné závěry nicméně přinesou až rozsáhlejší metaanalýzy a studie typu „head to head“.
Seznam použité literatury
- [1] Rask‑Andersen M, Zhang J, Fabbro D. Advances in kinase targeting: current clinical use and clinical trials. Trends Pharmacol Sci 2014; 35: 604−620.
- [2] Faivre S, Djelloul S, Raymond E. New paradigms in anticancer therapy: targeting multiple signaling pathways with kinase inhibitors. Amsterdam: Seminars in oncology Elsevier; 2006: 407–420.
- [3] Collett MS, Erikson R. Protein kinase activity associated with the avian sarcoma virus src gene product. Proc Natl Acad Sci 1978; 75: 2021–2024.
- [4] Castagna M, Takai Y, Kaibuchi K. Direct activation of calcium‑activated, phospholipid‑dependent protein kinase by tumor‑promoting phorbol esters. J Biol Chem 1982; 257: 7847–7851.
- [5] Lozzio CB, Lozzio BB. Human chronic myelogenous leukemia cell‑line with positive Philadelphia chromosome. Blood 1975; 45: 321–334.
- [6] Schultz KR, Bowman WP, Aledo A. Improved early event‑free survival with imatinib in Philadelphia chromosome–positive acute lymphoblastic leukemia: a children's oncology group study. J Clin Oncol 2009; 27: 5175–5181.
- [7] Coussens L, Parker PJ, Rhee L. Multiple, distinct forms of bovine and human protein kinase C suggest diversity in cellular signaling pathways. Science 1986; 233: 859–867.
- [8] Manning G, Whyte DB, Martinez R. The protein kinase complement of the human genome. Science 2002; 298: 1912–1934.
- [9] Fabbro D, Cowan‑Jacob SW, Moebitz H. Ten things you should know about protein kinases: IUPHAR review 14. Br J Pharmacol 2015; 172: 2675–2700.
- [10] Köstler WJ, Zielinski CC. Targeting Receptor Tyrosine Kinases in Cancer. In Receptor Tyrosine Kinases: Structure, Functions and Role in Human Disease. New York: Spring; 2015: 225–278.
- [11] Maurer G, Tarkowski B, Baccarini M. Raf kinases in cancer‑roles and therapeutic opportunities. Oncogene 2011; 30: 3477–3488.
- [12] Sato S, Sanjo H, Takeda K. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat Immunol 2005; 6: 1087–1095.
- [13] Mueller BK, Mack H, Teusch N. Rho kinase, a promising drug target for neurological disorders. Nat Rev Drug Discov 2005; 4: 387–398.
- [14] Chong ZZ, Shang YC, Wang S. A critical kinase cascade in neurological disorders: PI3K, Akt and mTOR. Future Neurol 2012; 7: 733–748.
- [15] Tabit CE, Shenouda SM, Holbrook M. Protein kinase C‑β contributes to impaired endothelial insulin signaling in humans with diabetes mellitus. Circulation 2013; 127: 86–95.
- [16] Bhullar KS, Lagarón NO, McGowan EM. Kinase‑targeted cancer therapies: progress, challenges and future directions. Mol Cancer 2018; 17: 48.
- [17] Hilton JF, Shapiro GI. Aurora kinase inhibition as an anticancer strategy. J Clin Oncol 2014; 32: 57–59.
- [18] Ahmed K, Unger G, Kren BT. Targeting CK2 for cancer therapy using a nanomedicine approach. In Protein kinase CK2 cellular function in normal and disease states. New York: Springer; 2015: 299–315.
- [19] Cance WG, Kurenova E, Marlowe T. Disrupting the scaffold to improve focal adhesion kinase–targeted cancer therapeutics. Sci Signal 2013; 6: pe10.
- [20] Fayard E, Xue G, Parcellier A. Protein kinase B (PKB/ Akt), a key mediator of the PI3K signaling pathway. In: Phosphoinositide 3‑ kinase in health and disease. New York: Springer, 2011; 31–56.
- [21] Weiß L, Efferth T. Polo‑like kinase 1 as target for cancer therapy. Exp Hematol Oncol 2012; 1: 38.
- [22] Krisenko MO, Geahlen RL. Calling in SYK: SYK’s dual role as a tumor promoter and tumor suppressor in cancer. Biochimi Biophys Acta 2015; 1853: 254–263.
- [23] Knighton DR, Zheng J, Ten Eyck LF. Crystal structure of the catalytic subunit of cyclic adenosine monophosphate‑dependent protein kinase. Science 1991; 253: 407–414.
- [24] Zheng J, Knighton DR, Taylor SS. Crystal structures of the myristylated catalytic subunit of cAMP‑dependent protein kinase reveal open and closed conformations. Protein Sci 1993; 2: 1559−1573.
- [25] Karaman MW, Herrgard S, Treiber DK. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol 2008; 26: 127–132.
- [26] Shukla S, Robey RW, Bates SE. Sunitinib (Sutent, SU11248), a small‑molecule receptor tyrosine kinase inhibitor, blocks function of the ATP‑binding cassette (ABC) transporters P‑glycoprotein (ABCB1) and ABCG2. Drug Metab Dispos 2009; 37: 359–365.
- [27] Kirkland LO, McInnes C. Non‑ATP competitive protein kinase inhibitors as anti‑tumor therapeutics. Biochem Pharmacol 2009; 77: 1561–1571.
- [28] Mahon FX. Pharmacologic monitoring and determinants of intracytoplasmic drug level. Best Pract Res Clin Haematol 2009; 22: 381−386.
- [29] Nambu T, Hamada A, Nakashima R. Association of SLCO1B3 polymorphism with intracellular accumulation of imatinib in leukocytes in pacients with chronic myeloid leukemia. Biol Pharm Bull 2011; 34: 114−119.
- [30] White DL, Eadie LN, Saunders VA. Proton pump inhibiotors significantly increase the intracellular concentration of nilotinib, but not imatinib in target CML cell. Leukemia 2013; 27: 1201−12024.
- [31] Bouchet S, Dulucq S, Pasquet JM. From in vitro to in vivo: intracellular determination of imatinib and nilotinib may be related with clinical outcome. Leukemia 2013; 27: 1757−1759.
- [32] Drenberg CD, Baker SD, Sparreboom A. Integrating clinical pharmacology concepts in individualized therapy with tyrosine kinase inhibitors. Clin Pharmacol Ther 2013; 93: 215−219.
- [33] Josephs DH, Fisher DS, Spicer J. Clinical pharmocokinetics of tyrosine kinase inhibitors: implications for therapeutic drug monitoring. Ther Drug Monit 2013; 35: 562−587.
- [34] de Wit D, Guchelaar HJ, den Hartigh J. Individualized dosing of tyrosine kinase inhibitors: are we there yet? Drug Discov Today 2015; 20: 18−36.
- [35] Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol 2006; 2: 358–364.
- [36] Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 2009; 9: 28–39.
- [37] Force T, Kolaja KL. Cardiotoxicity of kinase inhibitors: the prediction and translation of preclinical models to clinical outcomes. Nat Rev Drug Discov 2011; 10: 111–126.
- [38] Hasinoff BB. The cardiotoxicity and myocyte damage caused by small molecule anticancer tyrosine kinase inhibitors is correlated with lack of target specificity. Toxicol Appl Pharmacol 2010; 244: 190–195.
- [39] Kufareva I, Abagyan R. Type‑II kinase inhibitor docking, screening, and profiling using modified structures of active kinase states. J Med Chem 2008; 51: 7921–7932.
- [40] Lamba V, Ghosh I. New directions in targeting protein kinases: focusing upon true allosteric and bivalent inhibitors. Curr Pharm Des 2012; 18: 2936–2945.
- [41] Eglen R, Reisine T. Drug discovery and the human kinome: recent trends. Pharmacol Ther 2011; 130: 144–156.
- [42] Blanc J, Geney R, Menet C. Type II kinase inhibitors: an opportunity in cancer for rational design. Anti‑Cancer Agents Med Chem (Formerly Current Medicinal Chemistry‑Anti‑Cancer Agents) 2013; 13: 731–747.
- [43] Cohen MS, Zhang C, Shokat KM. Structural bioinformatics‑based design of selective, irreversible kinase inhibitors. Science 2005; 308: 1318–1321.
- [44] Kwak EL, Sordella R, Bell DW. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci USA 2005; 102: 7665–7670.
- [45] Zhao Z, Liu Q, Bliven S. Determining cysteines available for covalent inhibition across the human kinome. J Med Chem 2017; 60: 2879–2889.
- [46] Baker SD, Hu S. Pharmacokinetics considerations for new targeted therapies. Clin Pharmacol Ther 2009; 85: 208−211.
- [47] Herbrink M, Nuijen B, Schellens JH. Variability in bioavailability of small molecular tyrosine kinase inhibitors. Cancer Treat Rev 2015; 41: 412–422.
- [48] Yu H, Steeghs N, Nijenhuis CM. Practical guidelines for therapeutic drug monitoring of anticancer tyrosine kinase inhibitors: focus on the pharmacokinetic targets. Clin Pharmacokinet 2014; 53: 305–325.
- [49] Widmer N, Bardin C, Chatelut E. Review of therapeutic drug monitoring of anticancer drugs part two – targeted therapies. Eur J Cancer 2014; 50: 2020–2036.
- [50] Terada T, Noda S, Inui K. Management of dose variability and side effects for individualized cancer pharmacotherapy with tyrosine kinase inhibitors. Pharmacol Ther 2015; 152: 125–134.
- [51] Wulkersdorfer B, Zeitlinger M, Schmid M. Pharmacokinetic aspects of vas‑cular endothelial growth factor tyrosine kinase inhibitors. Clin Pharmacokinet 2016; 55: 47–77.
- [52] Neul C, Schaeffeler E, Sparreboom A. Impact of Membrane Drug Transporters on Resistance to Small‑Molecule Tyrosine Kinase Inhibitors, Trends Pharmacol Sci 2016; 37: 904−932.
- [53] Burger H, van Tol H, Boersma AW. Imatinib mesylate (STI571) is a substrate for the breast cancer resistance protein (BCRP)/ABCG2 drug pump. Blood 2004; 104: 2940−2942.
- [54] Gillet, JP, Gottesman MM. Mechanisms of multidrug resistance in cancer. Methods Mol Biol 2010; 596: 47−76.
- [55] Wolking S, Schaeffeler E, Lerche H. Impact of genetic polymorphisms of ABCB1 (MDR1, P‑glycoprotein) on drug disposition and potential clinical implications: update of the literature. Clin Pharmacokinet 2015; 54: 709–735.
- [56] Durmus S, Hendrikx JJ, Schinkel AH. Apical ABC transporters and cancer chemotherapeutic drug disposition. Adv Cancer Res 2015; 125: 1–41.
- [57] Giacomini KM, Huang SM, Tweedie DJ. Membrane transporters in drug development. Nat Rev Drug Discov 2010; 9: 215–236.
- [58] Mendes P, Oliver SG, Kell DB. Fitting transporter activities to cellular drug concentrations and fluxes: why the bumblebee can fly. Trends Pharmacol Sci 2015; 36: 710–723.
- [59] Matsson P, Fenu LA, Lundquist P. Quantifying the impact of transporters on cellular drug permeability. Trends Pharmacol Sci 2015; 36: 255–262.
- [60] Matsson P, Lundquist P, Artusson P. The need for speed‑kinetic limits of drug transporters. Trends Pharmacol Sci 2016; 37: 243–245.
- [61] Mendes P, Oliver SG, Kell DB. Response to ‘The Need for Speed’, by Matsson et al. Trends Pharmacol Sci 2016; 37: 245–246.
- [62] Souhrn údajů o přípravku Ibrance, revize 24. 8. 2018. Dostupné na: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‑_Product_Information/human/003853/WC500217196.pdf
- [63] Touma JA, McLachlan AJ, Gross AS. The role of ethnicity in personalized dosing of small molecule tyrosine kinase inhibitors used in oncology; Transl Cancer Res 2017; 6(Suppl 10): S1558−S1591.
- [64] Goodman VL, Rock EP, Dagher R. Approval summary: sunitinib for the treatment of imatinib refractory or intolerant gastrointestinal stromal tumors and advanced renal cell carcinoma. Clin Cancer Res 2007; 13: 1367−1373.
- [65] Gschwind HP, Pfaar U, Waldmeier F. Metabolism and disposition of imatinib mesylate in healthy volunteers. Drug Metab Dispos 2005; 33: 1503−1512.
- [66] O'Donnell PH, Dolan ME. Cancer pharmacoethnicity: ethnic differences in susceptibility to the effects of chemotherapy. Clin Cancer Res 2009; 15: 4806−4814.
- [67] Patel JN. Cancer pharmacogenomics: implications on ethnic diversity and drug response. Pharmacogenet Genomics 2015; 25: 223−230.
- [68] Fukuoka M, Yano S, Giaccone G. Multi‑institutional randomized phase II trial of gefitinib for previously treated patients with advanced non‑small‑cell lung cancer (The IDEAL 1 Trial) [corrected]. J Clin Oncol 2003; 21: 2237−2246.
- [69] Nishiwaki Y, Yano S, Tamura T. Subset analysis of data in the Japanese patients with NSCLC from IDEAL 1 study on gefitinib. Gan To Kagaku Ryoho 2004; 31: 567–573.
- [70] Fiala O, Šatánková M, Kultan J. Výskyt mutací genu EGFR u pacientů s NSCL v České republice. Onkologie 2014; 8: 156−159.
- [71] Schabath MB, Cress WD, Munoz‑Antonia T. Racial and Ethnic Differences in the Epidemiology of Lung Cancer and Lung Cancer Genome. Cancer Control 2016; 23: 338−346.
- [72] Pao W, Miller V, Zakowski M. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A 2004; 101: 13306−13311.
- [73] Paez JG, Janne PA, Lee JC. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004; 304: 1497−1500.
- [74] Mok T, Wu YL, Au JS. Efficacy and safety of erlotinib in 1242 East/South‑East Asian patients with advanced non‑small cell lung cancer. J Thorac Oncol 2010; 5: 1609−1615.
- [75] Cappuzzo F, Ciuleanu T, Stelmakh L. Erlotinib as maintenance treatment in advanced non‑small‑cell lung cancer: a multicentre, randomised, placebo‑controlled phase 3 study. Lancet Oncol 2010; 11: 521−529.
- [76] Wu YL, Kim JH, Park K. Efficacy and safety of maintenance erlotinib in Asian patients with advanced non‑small‑cell lung cancer: a subanalysis of the phase III, randomized SATURN study. Lung Cancer 2012; 77: 339−345.
- [77] Shepherd FA, Rodrigues Pereira J. Erlotinib in previously treated non‑small‑cell lung cancer. N Engl J Med 2005; 353: 123−132.
- [78] Pilotto S, Di Maio M, Peretti U. Predictors of outcome for patients with lung adenocarcinoma carrying the epidermal growth factor receptor mutation receiving 1st‑line tyrosine kinase inhibitors: Sensitivity and metaregression analysis of randomized trials. Crit Rev Oncol Hematol 2014; 90: 135−145.
- [79] Chang A, Parikh P, Thongprasert S. Gefitinib (IRESSA) in patients of Asian origin with refractory advanced non‑small cell lung cancer: subset analysis from the ISEL study. J Thorac Oncol 2006; 1: 847−855.
- [80] Thatcher N, Chang A, Parikh P. Gefitinib plus best supportive care in previously treated patients with refractory advanced non‑small‑cell lung cancer: results from a randomised, placebo‑controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet 2005; 366: 1527−1537.
- [81] Guan ZZ, Zhang L, Li LY. Efficacy of gefitinib on Chinese patients with locally advanced or metastatic non‑small cell lung cancer: a clinical trial. Ai Zheng 2005; 24: 980−984.
- [82] Sherry ST, Ward M, Kholodov M. dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res 1999; 9: 677−679.
- [83] Kim DH, Sriharsha L, Xu W. Clinical relevance of a pharmacogenetic approach using multiple candidate genes to predict response and resistance to imatinib therapy in chronic myeloid leukemia. Clin Cancer Res 2009; 15: 4750−4758.
- [84] Tamura M, Kondo M, Horio M. Genetic polymorphisms of the adenosine triphosphate‑binding cassette transporters (ABCG2, ABCB1) and gefitinib toxicity. Nagoya J Med Sci 2012; 74: 133−140.
- [85] Takahashi N, Miura M, Scott SA. Influence of CYP3A5 and drug transporter polymorphisms on imatinib trough concentration and clinical response among patients with chronic phase chronic myeloid leukemia. J Hum Genet 2010; 55: 731−737.
- [86] Petain A, Kattygnarath D, Azard J. Population pharmacokinetics and pharmacogenetics of imatinib in children and adults. Clin Cancer Res 2008; 14: 7102−7109.
- [87] Imai Y, Nakane M, Kage K. C421A polymorphism in the human breast cancer resistance protein gene is associated with low expression of Q141K protein and lowlevel drug resistance. Mol Cancer Ther 2002; 1: 611−616.
- [88] Koo DH, Ryu MH, Ryoo BY. Association of ABCG2 polymorphism with clinical efficacy of imatinib in patients with gastrointestinal stromal tumor. Cancer Chemother Pharmacol 2015; 75: 173−182.
- [89] Kim HR, Park HS, Kwon WS. Pharmacogenetic determinants associated with sunitinib‑induced toxicity and ethnic difference in Korean metastatic renal cell carcinoma patients. Cancer Chemother Pharmacol 2013; 72: 825−835.
- [90] Miura Y, Imamura CK, Fukunaga K. Sunitinib induced severe toxicities in a Japanese patient with the ABCG2 421 AA genotype. BMC Cancer 2014; 14: 964.
- [91] Fukudo M, Ikemi Y, Togashi Y. Population pharmacokinetics/pharmacodynamics of erlotinib and pharmacogenomic analysis of plasma and cerebrospinal fluid drug concentrations in Japanese patients with non‑small cell lung cancer. Clin Pharmacokinet 2013; 52: 593−609.
- [92] Cusatis G, Gregorc V, Li J. Pharmacogenetics of ABCG2 and adverse reactions to gefitinib. J Natl Cancer Inst 2006; 98: 1739−1742.
- [93] Li J, Cusatis G, Brahmer J. Association of variant ABCG2 and the pharmacokinetics of epidermal growth factor receptor tyrosine kinase inhibitors in cancer patients. Cancer Biol Ther 2007; 6: 432−438.
- [94] ČSÚ − R04 Cizinci v ČR podle státního občanství v letech 1994−2017 (k 31. 12. 2017).
- [95] Feely J, Grimm T. A comparison of drug protein binding and alpha 1‑acid glycoprotein concentration in Chinese and Caucasians. Br J Clin Pharmacol 1991; 31: 551−552.
- [96] Bins S, Eechoute K, Kloth JS. Prospective Analysis in GIST Patients on the Role of Alpha‑1 Acid Glycoprotein in Imatinib Exposure. Clin Pharmacokinet 2017; 56: 305−310.
- [97] Gambacorti‑Passerini C, Zucchetti M, Russo D. Alpha1 acid glycoprotein binds to imatinib (STI571) and substantially alters its pharmacokinetics in chronic myeloid leukemia patients. Clin Cancer Res 2003; 9: 625−632.
- [98] Zhong JS, Meng FY, Xu D. Serum alpha1‑acid glycoprotein, imatinib concentration and efficacy in chronic myeloid leukemia patients. Zhonghua Yi Xue Za Zhi 2011; 91: 2120−2123.
- [99] Wang J, Thornton JC, Russell M. Asians have lower body mass index (BMI) but higher percent body fat than do whites: comparisons of anthropometric measurements. Am J Clin Nutr 1994; 60: 23−28.
- [100] Deurenberg P, Deurenberg‑Yap M, Guricci S. Asians are different from Caucasians and from each other in their body mass index/body fat per cent relationship. Obes Rev 2002; 3: 141−146.
- [101] Rush EC, Goedecke JH, Jennings C. BMI, fat and muscle differences in urban women of five ethnicities from two countries. Int J Obes (Lond) 2007; 31: 1232−1239.
- [102] Lear SA, Kohli S, Bondy GP. Ethnic variation in fat and lean body mass and the association with insulin resistance. J Clin Endocrinol Metab 2009; 94: 4696−4702.
- [103] Chuah CT, Nakamae H, Shen ZX. Efficacy and safety of dasatinib versus imatinib in the East Asian subpopulation of the DASISION trial of newly diagnosed chronic myeloid leukemia in chronic phase. Leuk Lymphoma 2014; 55: 2093−2100.
- [104] Kiyota N, Schlumberger M, Muro K. Subgroup analysis of Japanese patients in a phase 3 study of lenvatinib in radioiodine‑refractory differentiated thyroid cancer. Cancer Sci 2015; 106: 1714−1721.
- [105] Schlumberger M, Tahara M, Wirth LJ. Lenvatinib versus placebo in radioiodine‑refractory thyroid cancer. N Engl J Med 2015; 372: 621−630.
- [106] Guo J, Jin J, Huang Y. Comparison of PFS and safety for Asian compared to North American and European populations in the phase III trial of pazopanib versus sunitinib in patients with treatment‑naive RCC (COMPARZ). J Clin Oncol 2013; 31: 366.
- [107] Escudier B, Eisen T, Stadler WM. Sorafenib in advanced clear‑cell renal‑cell carcinoma. N Engl J Med 2007; 356: 125−134.
- [108] Kudo M, Lencioni R, Marrero JA. Regional differences in sorafenib‑treated patients with hepatocellular carcinoma: GIDEON observational study. Liver Int 2016; 36: 1196−1205.
- [109] Liu X, Fiocco M, Swen JJ. Assessment of ethnic differences in sunitinib outcome between Caucasian and Asian patients with metastatic renal cell carcinoma: a meta‑analysis. Acta Oncol 2017; 56: 582−589.
- [110] Naito S, Tsukamoto T, Murai M. Overall survival and good tolerability of long‑term use of sorafenib after cytokine treatment: final results of a phase II trial of sorafenib in Japanese patients with metastatic renal cell carcinoma. BJU Int 2011; 108: 1813−1819.
- [111] Meijerman I, Beijnen JH, Schellens JH. Herb‑drug interactions in oncology: focus on mechanisms of induction. Oncologist 2006; 11: 742−752.
- [112] Strizich G, Gammon MD, Jacobson JS. Latent class analysis suggests four distinct classes of complementary medine user among women with breat cancer. BMC Complemet Alter Med 2015: 15: 411.
- [113] Lee AH, Ingraham SE, Kopp M. The incidence of potential interactions between dietary supplements and prescription medications in cancer patients at a Veterans Administration Hospital. Am J Clin Oncol 2006; 29: 178–182.
- [114] Halámková J. Vybrané interkace preparátů užívaných v onkologii s potravinovými doplňky a fytofarmaky – pohled onkologa. Ústní sdělení. XL. brněnské onkologické dny a XXX. konference pro nelékařské zdravotnické pracovníky, 2016.
- [115] Hsiao AF, Wong MD, Goldstein MS. Variation in complementary and alternative medicine (CAM) use across racial/ethnic groups and the development of ethnic‑specific measures of CAM use. J Altern Complement Med 2006; 12: 281−290.
- [116] Arcury TA, Suerken CK, Grzywacz JG. Complementary and alternative medicine use among older adults: ethnic variation. Ethn Dis 2006; 16: 723−731.
- [117] Gan GG, Leong YC, Bee PC. Complementary and alternative medicine use in patients with hematological cancers in Malaysia. Support Care Cancer 2015; 23: 2399−2406.
- [118] Sparber A, Bauer L, Curt G. Use of complementary medicine by adult patients participating in cancer clinical trials. Oncol Nurs Forum 2000; 27: 623−630.
- [119] Molassiotis A, Fernadez‑Ortega P, Pud D. Use of complementary and alternative medicine in cancer patients: a European survey. Ann Oncol 2005; 16: 655−663.
- [120] Kim MJ, Lee SD, Kim DR. Use of complementary and alternative medicine among Korean cancer patients. Korean J Intern Med 2004; 19: 250−256.
- [121] SZÚ – Užívání tabáku v České republice 2015. Dostupné na: www.szu.cz/uploads/documents/czzp/zavislosti/Uzivani_tabaku_2015.pdf
- [122] Asma S, Mackay J, Song SY, on behalf of the GATS Collaborative Group. The GATS Atlas. CDC Foundation, Atlanta, 2015.
- [123] Faber MS, Fuhr U. Time response of cytochrome P450 1A2 activity on cessation of heavy smoking. Clin Pharmacol Ther 2004; 76: 178−184.
- [124] Hamilton M, Wolf JL, Rusk J. Effects of smokingon the pharmacokinetics of erlotinib. Clin Cancer Res 2006; 12: 2166−2171.
- [125] Hamilton M, Wolf JL, Zborowski D. Tarceva (erlotinib) exposure/effects (EE) analysis from a Phase III study in advanced NSCLC: Effect of smoking on the PKof erlotinib. Cancer Res 2005; 65: Abstract 6165.
- [126] Zhang Y, Kang S, Fang W. Impact of smoking status on EGFR‑TKI efficacy for advanced non‑small‑cell lung cancer in EGFR mutants: a meta‑analysis. Clin Lung Cancer 2015; 16: 144−151.
- [127] Faderl S, Talpaz M, Estrov Z. Chronic myelogenous leukemia: biology and therapy. Ann Intern Med 1999; 131: 207–219.
- [128] Au WY, Caguioa PB, Chuah C. Chronic myeloid leukemia in Asia. Int J Hematol 2009; 89: 14−23.
- [129] Kim DH, Kong JH, Byeun JY. The IFNG (IFN‑gamma) genotype predicts cytogenetic and molecular response to imatinib therapy in chronic myeloid leukemia. Clin Cancer Res 2010; 16: 5339−5350.
- [130] Nakanishi T, Ross DD. Breast cancer resistance protein (BCRP/ABCG2): its role in multidrug resistence and regulation of its gene expression. Chin J Cancer 2012; 31: 73−99.
- [131] Takahashi N, Wakita H, Miura M. Correlation between imatinib pharmacokinetics and clinical response in Japanese patients with chronic‑phase chronic myeloid leukemia. Clin Pharmacol Ther 2010; 88: 809−813.
- [132] Hodges LM, Markova SM, Chinn LW. Very important pharmacogene summary: ABCB1 (MDR1, P‑glycoprotein). Pharmacogenet Genomics 2011; 21: 152−161.
- [133] Zheng Q, Wu H, Yu Q. ABCB1 polymorphisms predict imatinib response in chronic myeloid leukemia patients: a systematic review and meta‑analysis. Pharmacogenomics J 2015; 15: 127−134.
- [134] Nakamae H, Fujisawa S, Ogura M. Dasatinib versus imatinib in Japanese patients with newly diagnosed chronic phase chronic myeloid leukemia: a subanalysis of the DASISION 5‑year final report. Int J Hematol 2017; 105: 792−804.
- [135] Saglio G, Kim DW, Issaragrisil S. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med 2010; 362: 2251−2259.
- [136] Nakamae H, Shibayama H, Kurokawa M. Nilotinib as frontline therapy for patients with newly diagnosed Ph+ chronic myeloid leukemia in chronic phase: results from the Japanese subgroup of ENESTnd. Int J Hematol 2011; 93: 624−632.
- [137] Wang J, Shen ZX, Saglio G. Phase 3 study of nilotinib vs imatinib in Chinese patients with newly diagnosed chronic myeloid leukemia in chronic phase: ENESTchina. Blood 2015; 125: 2771−2778.
- [138] Cortes JE, Kim DW, Pinilla‑Ibarz J. A phase 2 trial of ponatinib in Philadelphia chromosome‑positive leukemias. N Engl J Med 2013; 369: 1783−1796.
- [139] Tojo A, Kyo T, Yamamoto K. Ponatinib in Japanese patients with philadelphia chromosome‑positive leukemia, a phase 1/2 study Int J Hematol 2017; 106: 385−397.
- [140] Cortes JE, Kantarjian HM, Brummendorf TH. Safety and efficacy of bosutinib (SKI‑606) in chronic phase Philadelphia chromosome‑positive chronic myeloid leukemia patients with resistance or intolerance to imatinib. Blood 2011; 118: 4567−4576.
- [141] Nakaseko C, Takahashi N, Ishizawa K. A phase 1/2 study of bosutinib in Japanese adults with Philadelphia chromosome‑positive chronic myeloid leukemia. Int J Hematol 2015; 101: 154−164.
- [142] Hsyu PH, Gogat K, Duvillie L. Pharmacokinetics and tolerability of bosutinib in Asian versus Non‑Asian patients with philadelphia chromosome-positive leukemia. Blood 2012; 120: 4440.
- [143] Yu Y, He J. Molecular classification of non‑small‑cell lung cancer: diagnosis, individualized treatment, and prognosis. Front Med 2013; 7: 157−171.
- [144] Savas P, Hughes B, Solomon B. Targeted therapy in lung cancer: IPASS and beyond, keeping abreast of the explosion of targeted therapies for lung cancer. J Thorac Dis 2013; 5(Suppl 5): S579–S592.
- [145] Shigematsu H, Lin L, Takahashi T. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 2005; 97: 339−346.
- [146] Shi L, Tang J, Tong L. Risk of interstitial lung disease with gefitinib and erlotinib in advanced non‑small cell lung cancer: a systematic review and meta‑analysis of clinical trials. Lung Cancer 2014; 83: 231−239.
- [147] Bell DW, Brannigan BW, Matsuo K. Increased prevalence of EGFR‑mutant lung cancer in women and in East Asian populations: analysis of estrogen‑related polymorphisms. Clin Cancer Res 2008; 14: 4079−4084.
- [148] Wang JP, Wu CY, Yeh YC. Erlotinib is effective in pancreatic cancer with epidermal growth factor receptor mutations: a randomized, open‑label, prospective trial. Oncotarget 2015; 6: 18162−18173.
- [149] Phelps MA, Stinchcombe TE, Blachly JS. Erlotinib in African Americans with advanced non‑small cell lung cancer: a prospective randomized study with genetic and pharmacokinetic analyses. Clin Pharmacol Ther 2014; 96: 182−191.
- [150] Fung KL, Gottesman MM. A synonymous polymorphism in a common MDR1 (ABCB1) haplotype shapes protein function. Biochim Biophys Acta 2009; 1794: 860−871.
- [151] Chu YH, Li H, Tan HS. Association of ABCB1 and FLT3 polymorphisms with toxicities and survival in asian patients receiving sunitinib for renal cell carcinoma. PLoS One 2015; 10: e0134102.
- [152] Hamada A, Sasaki J, Saeki S. Association of ABCB1 polymorphisms with erlotinib pharmacokinetics and toxicity in Japanese patients with non‑small‑cell lung cancer. Pharmacogenomics 2012; 13: 615−624.
- [153] Kai Y, Hamada A, Sasaki Ji. Association of CYP1A1 and CYP3A5 polymorphisms with pharmacokinetics of erlotinib in patients with non‑small cell lung cancer. Cancer Res 2011; 71(Suppl): Abstract 5459.
- [154] Kato T, Yoshioka H, Okamoto I. Afatinib versus cisplatin plus pemetrexed in Japanese patients with advanced non‑small cell lung cancer harboring activating EGFR mutations: Subgroup analysis of LUX‑Lung 3. Cancer Sci 2015; 106: 1202−1211.
- [155] Wu YL, Sequist LV, Schuler M. Overall survival with afatinib versus chemotherapy in patients with NSCLC harboring common EGFR mutations: subgroup analyses by race/ethnicity in LUX‑Lung 3 and LUX‑Lung 6. ESMO Asia 2015; 445P.
- [156] Wu YL, Cheng Y, Zhou X. Dacomitinib versus gefitinib as first‑line treatment for pacients with EGFR‑mutation‑positive non‑small‑cell lung cancer (ARCHER 1050): a randomised, open‑label, phase 3 trial. Lancet Oncol 2017; 18: 1454−1466.
- [157] Soria JC, Ohe Y, Vansteenkiste J. Osimertinib in Untreated EGFR‑Mutated Advanced Non‑Small‑Cell Lung Cancer. N Engl J Med 2018; 378: 113−125.
- [158] Awad MM, Shaw AT. ALK inhibitors in non‑small cell lung cancer: crizotinib and beyond. Clin Adv Hematol Oncol 2014; 12: 429–439.
- [159] Shaw AT, Kim DW, Mehra R. Ceritinib in ALK‑rearranged non‑small‑cell lung cancer. N Engl J Med 2014; 370: 1189–1197.
- [160] Schabath MB, Welsh EA, Fulp WJ. Differential association of STK11 and TP53 with KRAS mutation‑associated gene expression, proliferation and immune surveillance in lung adenocarcinoma. Oncogene 2016; 35: 3209−3216.
- [161] Gao B, Sun Y, Zhang J. Spectrum of LKB1, EGFR, and KRAS mutations in Chinese lung adenocarcinomas. J Thorac Oncol 2010; 5: 1130−1135.
- [162] Nishio M, Hirsh V, Kim DW. Efficacy, safety, and patient‑reported outcomes (PROs) with crizotinib versus chemotherapy in Asian patients in a phase III study of previously treated advanced ALK positive non‑small cell lung cancer. J Thorac Oncol 2013; 8: 2818.
- [163] Tan DS, Shaw AT, Mehra R. Ceritinib in Asian versus Caucasian patients with advanced anaplastic lymphoma kinase (ALK)‑rearranged (ALK+) NSCLC: Subgroup analysis of the ASCEND‑1 trial. J Clin Oncol 2014; 32: Abstract 8078.
- [164] Hida T, Shi Y, Ahn MJ. Exploratory subgroup analysis of crizotinib efficacy and safety in Asian and non‑Asian patients with advanced ALK‑positive non‑small cell lung cancer (NSCLC) enrolled in a global phase II study. J Thorac Oncol 2012; 7: 5.
- [165] Reck M, Kaiser R, Mellemgaard A. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non‑small‑cell lung cancer (LUME‑Lung 1):a phase 3, double‑blind, randomised controlled trial. Lancet Oncol 2014; 15: 143−155.
- [166] Okamoto I, Kaneda H, Satoh T. Phase I safety, pharmacokinetic, and biomarker study of BIBF 1120, an oral triple tyrosine kinase inhibitor in patients with advanced solid tumors. Mol Cancer Ther 2010; 9: 2825−2833.
- [167] Mross K, Stefanic M, Gmehling D. Phase I study of the angiogenesis inhibitor BIBF 1120 in patients with advanced solid tumors. Clin Cancer Res 2010; 16: 311−319.
- [168] Lee SH, Bang YJ, Mainwaring P. Sunitinib in metastatic renal cell carcinoma: an ethnic Asian subpopulation analysis for safety and efficacy. Asia Pac J Clin Oncol 2014; 10: 237−245.
- [169] Zhang Y, Mai H, Guo G. Association analysis of SNPs present in plasma with adverse events and population pharmacokinetics in Chinese sunitinib treated patients with renal cell carcinoma. Oncotarget 2018; 9: 14109−14123.
- [170] Poprach A, Büchler T, Lakomý R. Cílená léčba metastatického renálního karcinomu, možnosti sekvenční léčby: současný pohled. Onkologie 2014; 8: 80–85.
- [171] Tan HS, Li H, Hong YW. Efficacy and safety of an attenuated‑dose sunitinib regimen in metastatic renal cell carcinoma: results from a prospective registry in singapore. Clin Genitourin Cancer 2015; 13: e285−e295.
- [172] Ye DW, Zhang LH. Critical appraisal of sorafenib in the treatment of Chinese patients with renal cell carcinoma. Onco Targets Ther 2014; 7: 925–935.
- [173] Rini BI, Escudier B, Tomczak P. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet 2011; 378: 1931−1939.
- [174] Qin S, Bi F, Jin J. Axitinib versus sorafenib as a second‑line therapy in Asian patients with metastatic renal cell carcinoma: results from a randomized registrational study. Onco Targets Ther 2015; 8: 1363−1373.
- [175] Ueda T, Uemura H, Tomita Y. Efficacy and safety of axitinib versus sorafenib in metastatic renal cell carcinoma: subgroup analysis of Japanese patients from the global randomized Phase 3 AXIS trial. Jpn J Clin Oncol 2013; 43: 616−628.
- [176] Escudier B, Eisen T, Stadler WM. Sorafenib in advanced clear‑cell renal‑cell carcinoma. N Engl J Med 2007; 356: 125−134.
- [177] Qin C, Cao Q, Li P. The influence of genetic variants of sorafenib on clinical outcomes and toxic effects in patients with advanced renal cell carcinoma. Scientific Reports 2016; 6: 20089.
- [178] Tomita Y, Fukasawa S, Oya M. Predictive factors for efficacy of axitinib in first‑line metastatic renal cell carcinoma: subgroup analysis in Japanese patients from a randomized, double‑blind phase II study. Jpn J Clin Oncol 2016; 46: 1031–1041.
- [179] Zhou L, Liu XD, Sun M. Targeting MET and AXL overcomes resistance to sunitinib therapy in renal cell carcinoma. Oncogene 2016; 35: 2687–2697.
- [180] Motzer RJ, Hutson TE, Glen H. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open‑label, multicentre trial. Lancet Oncol 2015; 16: 1473−1482.