Farmakogenomika hypolipidemik
Dyslipidemie je jedním ze zásadních a současně modifikovatelných rizikových faktorů kardiovaskulárních onemocnění. Hypolipidemika, zejména statiny spolu s fibráty, patří k nejčastěji podávaným lékům vůbec. Genetickou komponentou interindividuálních odlišností v odpovědi na terapii se zabývá farmakogenetika. Mezi kandidáty s důkazy pro modulaci účinků fibrátů se objevují genový klastr APOA1/C3/A4/A5, polymorfismy v genu pro lipoproteinovou lipázu, jaterní lipázu nebo varianty přímo v cílovém genu fibrátů (PPAR-? – peroxisome proliferator-activated receptor alpha). U ezetimibu, blokátoru absorpce cholesterolu enterocyty, jsou všechny doposud popsané farmakogenetické interakce závislé na polymorfismech v cílovém genu NPC1L1. U statinů je popsána celá řada genetických variant ovlivňujících farmakokinetické a farmakodynamické aspekty jejich účinku a nově jsou k dispozici výsledky sedmi celogenomových asociačních studií, které dokládají posun od farmakogenetiky k éře farmakogenomiky hypolipidemik.
Úvod
Dyslipidemie je považována za jeden ze zásadních a současně modifikovatelných rizikových faktorů kardiovaskulárních onemocnění [1]. Ačkoli jsou základem léčby dyslipidemií především režimová opatření, často je nutné tuto léčbu doplnit o farmakoterapii. Hypolipidemika, zejména statiny spolu s fibráty, patří k nejčastěji podávaným lékům vůbec. I přes prokázanou účinnost hypolipidemik v primární i sekundární prevenci kardiovaskulárních onemocnění je zřejmá interindividuální variabilita jejich účinku, která do značné míry závisí na genetické výbavě jedince. Cílem farmakogenetiky je vývoj individualizovaných léčebných postupů, a to především určení nejvhodnějšího typu léčiva a stanovení jeho dávkování pro konkrétního pacienta. Prvním krokem na této cestě je identifikace genetických variant zodpovědných za odlišnou reakci na určitý lék. Tato zkoumání se provádějí nejčastěji pomocí asociačních studií, které přímo testují asociaci mezi fenotypem, například nežádoucím účinkem léčiva, a zkoumanou genetickou variantou. Nejčastěji používaným designem jsou tzv. studie případů a kontrol (case-control studies), ve kterých porovnáváme frekvence polymorfismu(ů) mezi skupinou vykazující studovaný fenotyp a skupinou kontrolní, kam mohou patřit například jedinci léčení stejnou látkou, u kterých se neprojevil vedlejší účinek. Pokud je rozdíl frekvencí pro některý z polymorfismů statisticky signifikantní, lze mluvit o farmakogenetické interakci. Mezi nevýhody asociačních studií patří nutnost co nejlepší shody porovnávaných skupin v prakticky všech parametrech vyjma zkoumaného znaku (zastoupení mužů a žen, etnických skupin, porovnatelnost věku a klinických parametrů), aby v důsledku stratifikace nebylo dosaženo falešně pozitivního výsledku, který by se samotnou farmakogenetickou interakcí vůbec nesouvisel. Dalším limitujícím aspektem asociačních studií byla nutnost výběru „kandidátního“ polymorfismu a priori na základě známých chemických nebo fyziologických souvislostí. V současné době však asociační studie prožívají nebývalou renesanci v podobě tzv. celogenomových asociačních studií (GWAS – genome-wide association studies). Ačkoli základní princip asociace zůstává zachován, jsou vyhodnocovány stovky tisíc polymorfních markerů u všech případů a kontrol a následně, po odfiltrování falešně pozitivních výsledků, lze identifikovat ty genetické varianty, jejichž zastoupení se mezi oběma skupinami nejvíce odlišuje. Je nasnadě, že tyto farmakogenomické studie tedy mohou identifikovat zcela nové polymorfismy v DNA, asociované s mírou účinku zkoumaného léčiva nebo s nežádoucím účinkem.
Farmakogenomika a statiny
Účinek statinů je zprostředkován inhibicí klíčového enzymu biosyntézy cholesterolu 3-HMG-CoA reduktázy. Statiny jsou indikovány především u nemocných s izolovanou hypercholesterolemií, ale také u pacientů se smíšenou hyperlipidemií [1]. Ačkoli je bezpečnostní profil statinů velice dobrý, jejich masové používání zvyšuje pravděpodobnost „setkání“ statinu se specifickým zcitlivujícím genotypem a tím i pravděpodobnost rozvoje nežádoucích účinků, zejména myopatie. Farmakogenetické aspekty léčby statiny byly v posledních letech předmětem několika souhrnných sdělení (z českých např. [2]). Farmakokinetiku statinů výrazně ovlivňují polymorfismy jednak v genech k![Obr. 1 Kandidátní geny ovlivňující farmakokinetiku statinů; volně podle [31]. Perorálně užívané statiny vstupují do systémové cirkulace z enterocytu prostřednictvím aktivních i pasivních mechanismů. Hlavními orgány jejich metabolismu a eliminace jsou játra a částečně i ledviny. Transport statinů přes buněčnou membránu zajišťují membránové transportéry z rodiny SLC (solute carrier) či ABC (ATP-binding cassette). Na metabolismu statinů se podílejí izoformy cytochromu P450 (CYP) a glykosyltransferáza (UGT). Jednotlivé varianty se liší afinitou k jednotlivým statinům a jejich metabolitům. Vysvětlivky: fialová – léčivo nebo jeho metabolit, modrá – membránový přenašeč, růžová – enzym metabolismu (Copyright © 2012 PharmGKB. Použito se svolením PharmGKB a Stanford University.)](https://www.remedia.cz/photo-a-28923---.jpg) ódujících izoformy cytochromů P450, a sice CYP3A4 (metabolizující lovastatin, simvastatin, atorvastatin) a CYP2C9 (metabolizující fluvastatin), dále řada membránových transportérů, jako je P-glykoprotein kódovaný genem ABCB1, či zástupci rodin transportních peptidů organických aniontů (OATP – organic anion transporting polypeptide, kódované geny SLC22A) a další (obr. 1). Mezi nejvýznamnějšími kandidátními geny, které ovlivňují farmakodynamické aspekty účinku statinů, jsou uváděny především geny kódující apolipoprotein E a HMG-CoA reduktázu. Publikovány byly již i výsledky části farmakogenetické studie účinnosti statinů v České republice. Ve skupině pacientů léčených statiny (simvastatin 46,3 %, atorvastatin 40,5 %, lovastatin 13,2 %) bylo zjištěno, že ve smyslu snížení hladiny LDL cholesterolu mají z léčby menší přínos nositelé alely C-1131 genu pro apolipoprotein A5 [3].
ódujících izoformy cytochromů P450, a sice CYP3A4 (metabolizující lovastatin, simvastatin, atorvastatin) a CYP2C9 (metabolizující fluvastatin), dále řada membránových transportérů, jako je P-glykoprotein kódovaný genem ABCB1, či zástupci rodin transportních peptidů organických aniontů (OATP – organic anion transporting polypeptide, kódované geny SLC22A) a další (obr. 1). Mezi nejvýznamnějšími kandidátními geny, které ovlivňují farmakodynamické aspekty účinku statinů, jsou uváděny především geny kódující apolipoprotein E a HMG-CoA reduktázu. Publikovány byly již i výsledky části farmakogenetické studie účinnosti statinů v České republice. Ve skupině pacientů léčených statiny (simvastatin 46,3 %, atorvastatin 40,5 %, lovastatin 13,2 %) bylo zjištěno, že ve smyslu snížení hladiny LDL cholesterolu mají z léčby menší přínos nositelé alely C-1131 genu pro apolipoprotein A5 [3].
Zajímavá a do značné míry ilustrativní je situace okolo varianty rs20455, vedoucí k záměně aminokyseliny (Trp719Arg) v kinesinu podobném proteinu 6 (KIF6). Výsledky z několika randomizovaných klinických studií (CARE, WOSCOPS, PROSPER a PROVE IT-TIMI 22) ukazují, že nositelé alely KIF6 719Arg jsou terapií statiny výrazně chráněni před koronárními příhodami na rozdíl od jedinců, kteří tuto alelu nemají (zde nebyl zjištěn žádný ochranný efekt) [4]. Tyto výsledky ovšem byly zpochybněny daty ze studií JUPITER, HPS a nedávno i TNT a IDEAL, jejichž autoři nejen že nenašli souvislost mezi alelou KIF6 719Arg a kardiovaskulárním rizikem, ale nezjistili ani farmakogenetickou interakci mezi zmiňovanou alelou a statiny [5]. Příčin těchto diskrepantních nálezů může být celá řada: rozdílný design studií, použitý typ a dávky statinů, stratifikace kohort a jiné, nicméně většina z nich pravděpodobně tkví v základní premise, jíž je hledání „absolutních“ rizikových nebo ochranných genetických polymorfismů. Následné studie by tedy neměly odpovídat na otázku, která z obou stran sporu má pravdu, ale spíše by jejich pozornost měla být zaměřena na charakterizaci okolností (geoetnická skupina, věk, pohlaví, klinický a metabolický profil, typ a dávka statinu), za kterých má alela KIF6 719Arg nejsilnější predikční potenciál.
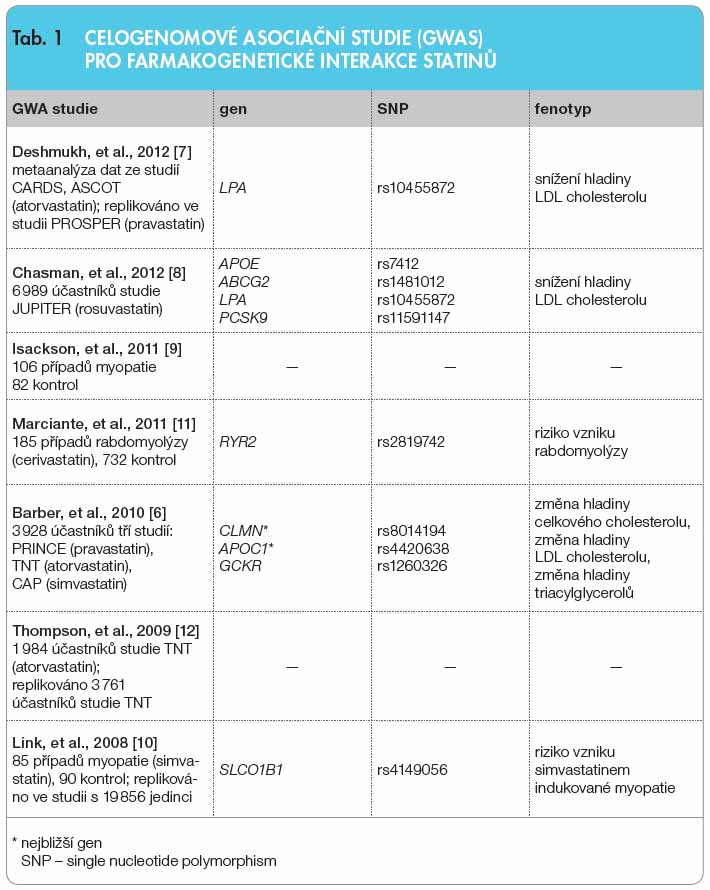 Oblastí, která zaznamenala zásadní rozvoj, je farmakogenomika léčby statiny, jejíž metodologickou část tvoří celogenomové asociační studie (GWAS). V současné době máme k dispozici výsledky sedmi studií GWAS [6–12]. V pěti z nich se autorům podařilo zjistit statisticky významné asociace vesměs jednonukleotidových polymorfismů, buďto s mírou statiny navozené změny lipidových parametrů, nebo přímo se svalovými komplikacemi léčby, jak ukazuje tab. 1. Kromě potvrzení některých polymorfismů, jejichž zapojení do farmakogenetických interakcí se statiny dokazovaly již studie kandidátních genů, přinesly GWAS řadu nových zajímavých kandidátů, jakými jsou například ryanodinový receptor 2 u cerivastatinem indukované rabdomyolýzy nebo opakovaně zjištěný jednonukleotidový polymorfismus rs10455872 v genu LPA.
Oblastí, která zaznamenala zásadní rozvoj, je farmakogenomika léčby statiny, jejíž metodologickou část tvoří celogenomové asociační studie (GWAS). V současné době máme k dispozici výsledky sedmi studií GWAS [6–12]. V pěti z nich se autorům podařilo zjistit statisticky významné asociace vesměs jednonukleotidových polymorfismů, buďto s mírou statiny navozené změny lipidových parametrů, nebo přímo se svalovými komplikacemi léčby, jak ukazuje tab. 1. Kromě potvrzení některých polymorfismů, jejichž zapojení do farmakogenetických interakcí se statiny dokazovaly již studie kandidátních genů, přinesly GWAS řadu nových zajímavých kandidátů, jakými jsou například ryanodinový receptor 2 u cerivastatinem indukované rabdomyolýzy nebo opakovaně zjištěný jednonukleotidový polymorfismus rs10455872 v genu LPA.
Farmakogenomika a fibráty
Fibráty jsou agonisté nukleárních receptorů PPAR (peroxisome proliferator- -activated receptor) [13], podávané u kombinovaných a hypertriglyceridemických forem hyperlipoproteinemií. Stejně jako u statinů jsou i v případě fibrátů nejčastěji popisovanými formami nežádoucích účinků myopatie [14]. Dosud nebyla provedena celogenomová, tedy farmakogenomická studie, která by se pokusila bez předchozí hypotézy (kandidátního genu) identifikovat polymorfismy ovlivňující míru hypolipidemického účinku fibrátů nebo senzitivitu k nežádoucím účinkům. Mezi kandidátními geny s důkazy pro modulaci účinků fibrátů se objevuje častěji například genový klastr APOA1/C3/A4/A5 na chromozomu 11 jako celek nebo varianty jeho jednotlivých členů [15–17], dále polymorfismy v genu pro lipoproteinovou lipázu [17], jaterní lipázu [18] nebo varianty přímo v cílovém genu fibrátů, PPAR-? [19]. U pacientů se smíšenou hyperlipoproteinemií je, i přes určité riziko vzniku komplikací (myopatie), indikována kombinační léčba statin + fibrát. Efekt polymorfismů v genu pro lipoproteinovou lipázu na hladinu apoC-III (apolipoproteinu C-III) byl zjištěn u 1250 takto léčených pacientů v rámci jedné z mála farmakogenetických studií. U nositelů haplotypu složeného z minoritních alel SNP rs1801177, rs7016529 a rs249 (frekvence haplotypu byla 2 %) došlo k paradoxnímu zvýšení hladiny apoC-III po kombinované terapii statin + fibrát. Autoři jako jeden z možných mechanismů uvádějí, že jimi identifikovaná genetická varianta vede k intenzivnější interakci lipoproteinové lipázy s apoC-III, která má za následek snížení její aktivity, tedy snížení clearance lipoproteinů bohatých na triacylglyceroly a zvýšení koncentrace apoC-III [20].
Farmakogenomika a ezetimib
U ezetimibu, blokátoru absorpce cholesterolu v membráně enterocytu, jsou informace o farmakogenomických i farmakogenetických aspektech doposud kusé a všechny směřují přímo k cílové molekule ezetimibu, NPC1L1. Ezetimib blokuje endocytózu NPC1L1 indukovanou steroly, a tím i absorpci cholesterolu (a fytosterolů). Vznikající chylomikrony jsou tak ochuzeny o značnou část cholesterolu, který transportují do jater. Snížený přísun cholesterolu do hepatocytů vede ke zvýšené expresi LDL receptorů, a tedy k intenzivnějšímu vychytávání aterogenních LDL a IDL částic z krve [21]. Ezetimib se používá i v monoterapii, častěji však v kombinaci se statiny při takzvané duální inhibici, tj. současné blokádě intestinální absorpce (ezetimib) a jaterní syntézy (statin) cholesterolu. První genetická studie u pacienta non-responzivního na ezetimib identifikovala vzácnou mutaci (složená heterozygocie V55L/I1233N) právě v genu pro NP1C1L1 [22], což vedlo k následné detailnější charakterizaci celého genu pomocí sekvenace. Jiná práce identifik![Obr. 2 Dvojrozměrný model membránové topologie lidského NPC1L1. Bílé a šedé obdélníky reprezentují transmembránové domény, šedé pak oblast citlivou na steroly (SSD, sterol-sensing domain). Černými značkami jsou označena místa mutací nalezená u jedinců s nízkou absorpcí cholesterolu [28]. Bílými značkami jsou označeny mutace ovlivňující účinek ezetimibu (viz text); podle [28] – Wang, et al., 2011.](https://www.remedia.cz/photo-a-28925---.jpg) ovala specifickou kombinaci tří jednonukleotidových polymorfismů (SNP), haplotyp 1735C-25342A-27677T, který významně měnil míru snížení hladiny LDL cholesterolu ezetimibem (větší efekt byl pozorován u jedinců, kteří tento běžný haplotyp neměli) [23]. Naopak, přímo u účastníků studií EASE a VYVA, kteří byli nositeli haplotypu -133A, -18A, 1679G, byla prokázána výraznější redukce hladiny LDL cholesterolu ezetimibem [24]. Následně byly objeveny další tři mutace u jedinců hyperresponzivních na účinky ezetimibu – R174H, 872CG (L272L) a 3929GA (Y1291Y) [25–27] (obr. 2). Není bez zajímavosti, že ani jedna z těchto variant není lokalizována v místě C-kličky NPC1L1, přímo zodpovědné za vazbu ezetimibu. Na svou první skutečně farmakogenomickou analýzu však ezetimib stále čeká.
ovala specifickou kombinaci tří jednonukleotidových polymorfismů (SNP), haplotyp 1735C-25342A-27677T, který významně měnil míru snížení hladiny LDL cholesterolu ezetimibem (větší efekt byl pozorován u jedinců, kteří tento běžný haplotyp neměli) [23]. Naopak, přímo u účastníků studií EASE a VYVA, kteří byli nositeli haplotypu -133A, -18A, 1679G, byla prokázána výraznější redukce hladiny LDL cholesterolu ezetimibem [24]. Následně byly objeveny další tři mutace u jedinců hyperresponzivních na účinky ezetimibu – R174H, 872CG (L272L) a 3929GA (Y1291Y) [25–27] (obr. 2). Není bez zajímavosti, že ani jedna z těchto variant není lokalizována v místě C-kličky NPC1L1, přímo zodpovědné za vazbu ezetimibu. Na svou první skutečně farmakogenomickou analýzu však ezetimib stále čeká.
Farmakogenomika a ostatní hypolipidemika
Kyselina nikotinová (niacin) je jedním z nejúčinnějších a nejdéle používaných hypolipidemik. Řada nežádoucích účinků spojených s jeho podáváním doposud bránila častějšímu terapeutickému použití. Ačkoli je znám hlavní mechanismus účinku niacinu zprostředkovaný s G-proteiny spřaženým receptorem GPR109A, zatím prakticky neexistují relevantní studie o farmakogenetických interakcích. Jednou z výjimek je relativně malá studie čínských dyslipidemických pacientů, u kterých polymorfismus v DGAT2 (diacylglycerol acyltransferáza 2) ovlivňoval míru snížení obsahu tuku v játrech [29]. Naopak mezi nejrecentnější přírůstky do rodiny hypolipidemik patří inhibitory CETP (cholesterol ester transfer protein). I u této skupiny se dá předpokládat významný přínos farmakogenetiky při vývoji a personalizaci léčby, zvláště po zastavení vývoje torcetrapibu na sklonku roku 2006 (především pro zvýšené riziko kardiovaskulárních a dalších komplikací) a dalcetrapibu v květnu 2012 (nedostatek klinicky významné účinnosti). Právě farmakogenetická metaanalýza čítající více než 80 000 osob zřetelně dokumentovala interakci mezi polymorfismy v genu pro CETP a účinkem torcetrapibu na lipidové parametry, nikoli však pro torcetrapibem indukované zvýšení krevního tlaku [30]. Na genetické úrovni se tak podařilo potvrdit „off-target“, tedy s inhibicí CETP nesouvisející původ nežádoucích účinků torcetrapibu, a byl položen základ pro testování a vývoj chemicky odlišných a bezpečnějších inhibitorů CETP.
Závěr
Závěrem je možné konstatovat, že se farmakogenetika a farmakogenomika hypolipidemik v posledních letech posunula od dílčích studií k celogenomovým a systematickým přístupům, od nichž lze v relativně blízké budoucnosti očekávat validované genetické markery pro skutečně personalizovanou léčbu dyslipidemie.
Tato práce byla podpořena projekty PRVOUK-P25/ LF1/2 a LK11217 (výsledky tohoto projektu byly získány za finančního přispění Ministerstva školství, mládeže a tělovýchovy v rámci účelové podpory programu výzkumu, vývoje a inovací NÁVRAT).
Seznam použité literatury
- [1] Češka R. Cholesterol a ateroskleróza, léčba dyslipidémií. Praha: Triton, 2005: 343.
- [2] Hubacek JA, Adamkova V, Zidkova K, et al. Statin pharmacokinetics. Vnitrni lekarstvi 2008; 54: 62–67.
- [3] Hubacek JA, Adamkova V, Prusikova M, et al. Impact of apolipoprotein A5 variants on statin treatment efficacy. Pharmacogenomics 2009; 10: 945–950.
- [4] Li Y, Iakoubova OA, Shiffman D, et al. KIF6 polymorphism as a predictor of risk of coronary events and of clinical event reduction by statin therapy. Am J Cardiol 2010; 106: 994–998.
- [5] Arsenault BJ, Boekholdt SM, Hovingh GK, et al. The 719Arg variant of KIF6 and cardiovascular outcomes in statin-treated, stable coronary patients of the treating to new targets and incremental decrease in end points through aggressive lipid- -lowering prospective studies. Circ Cardiovasc Genet 2012; 5: 51–57.
- [6] Barber MJ, Mangravite LM, Hyde CL, et al. Genome-wide association of lipid-lowering response to statins in combined study populations. PloS ONE 5; 2010: e9763.
- [7] Deshmukh HA, Colhoun HM, Johnson T, et al. Genome-wide association study of genetic determinants of LDL-c response to atorvastatin therapy: Importance of Lp(a). J Lipid Res 2012; 53: 1000–1011.
- [8] Chasman DI, Giulianini F, Macfadyen J, et al. Genetic Determinants of Statin Induced LDL-C Reduction: The JUPITER Trial. Circ Cardiovasc Genet 2012; 5: 257–264.
- [9] Isackson PJ, Ochs-Balcom HM, Ma C, et al. Association of common variants in the human eyes shut ortholog (EYS) with statin-induced myopathy: evidence for additional functions of EYS. Muscle Nerve 2011; 44: 531–538.
- [10] Link E, Parish S, Armitage J, et al. SLCO1B1 variants and statin-induced myopathy – a genome- -wide study. N Engl J Med 2008; 359: 789–799.
- [11] Marciante KD, Durda JP, Heckbert SR, et al. Cerivastatin, genetic variants, and the risk of rhabdomyolysis. Pharmacogenet Genomics 2011; 21: 280–288.
- [12] Thompson JF, Hyde CL, Wood LS, et al. Comprehensive whole-genome and candidate gene analysis for response to statin therapy in the Treating to New Targets (TNT) cohort. Circ Cardiovasc Genet 2009; 2: 173–181.
- [13] Seda O, Sedova L. Peroxisome proliferator-activated receptors as molecular targets in relation to obesity and type 2 diabetes. Pharmacogenomics 2007; 8: 587–596.
- [14] Wu J, Song Y, Li H, Chen J. Rhabdomyolysis associated with fibrate therapy: review of 76 published cases and a new case report. Eur J Clin Pharmacol 2009; 65: 1169–1174.
- [15] Lai CQ, Arnett DK, Corella D, et al. Fenofibrate effect on triglyceride and postprandial response of apolipoprotein A5 variants: the GOLDN study. Art Thromb Vasc Biol 2007; 27: 1417–1425.
- [16] Liu Y, Ordovas JM, Gao G, et al. Pharmacogenetic association of the APOA1/C3/A4/A5 gene cluster and lipid responses to fenofibrate: the genetics of lipid-lowering drugs and diet network study. Pharmacogenet Genomics 2009; 19: 161–169.
- [17] Chien KL, Lin YL, Wen HC, et al. Common sequence variant in lipoprotein lipase gene conferring triglyceride response to fibrate treatment. Pharmacogenomics 2009; 10: 267–276.
- [18] Cenarro A, Artieda M, Gonzalvo C, et al. Genetic variation in the hepatic lipase gene is associated with combined hyperlipidemia, plasma lipid concentrations, and lipid-lowering drug response. Am Heart J 2005; 150: 1154–1162.
- [19] Foucher C, Rattier S, Flavell DM, et al. Response to micronized fenofibrate treatment is associated with the peroxisome-proliferator-activated receptors alpha G/C intron7 polymorphism in subjects with type 2 diabetes. Pharmacogenetics 2004; 14: 823–829.
- [20] Brautbar A, Virani SS, Belmont J, et al. LPL gene variants affect apoC-III response to combination therapy of statins and fenofibric acid in a randomized clinical trial of individuals with mixed dyslipidemia. JLR 2012; 53: 556–560.
- [21] Vaverkova H. Dual inhibition of cholesterol using the drug combination ezetimibe/simvastatin? Vnitrni lekarstvi 2007; 53: 421–427.
- [22] Wang J, Williams CM, and Hegele RA. Compound heterozygosity for two non-synonymous polymorphisms in NPC1L1 in a non-responder to ezetimibe. Clin Genet 2005; 67: 175–177.
- [23] Hegele RA, Guy J, Ban MR, and Wang J. NPC1L1 haplotype is associated with inter-individual variation in plasma low-density lipoprotein response to ezetimibe. Lipids Health Dis 2005; 4: 16.
- [24] Simon JS, Karnoub MC, Devlin DJ, et al. Sequence variation in NPC1L1 and association with improved LDL-cholesterol lowering in response to ezetimibe treatment. Genomics 2005; 86: 648–656.
- [25] Weinglass AB, Kohler M, Schulte U, et al. Extracellular loop C of NPC1L1 is important for binding to ezetimibe. Proc Nat Acad Sci USA 2008; 105: 11140–11145.
- [26] Pisciotta L, Fasano T, Bellocchio A, et al. Effect of ezetimibe coadministered with statins in genotype- confirmed heterozygous FH patients. Atherosclerosis 2007; 194: e116–122.
- [27] Zhao HL, Houweling AH, Vanstone CA, et al. Genetic variation in ABC G5/G8 and NPC1L1 impact cholesterol response to plant sterols in hypercholesterolemic men. Lipids 2008; 43: 1155–1164.
- [28] Wang LJ, Wang J, Li N, et al. Molecular characterization of the NPC1L1 variants identified from cholesterol low absorbers. J Biol Chem 2011; 286: 7397–7408.
- [29] Hu M, Chu WC, Yamashita S, et al. Liver fat reduction with niacin is influenced by DGAT- 2polymorphisms in hypertriglyceridemic patients. J Lipid Res 2012; 53: 802–809.
- [30] Sofat R, Hingorani AD, Smeeth L, et al. Separating the mechanism-based and off-target actions of cholesteryl ester transfer protein inhibitors with CETP gene polymorphisms. Circulation 2010; 5; 121: 52–62.
- [31] http://www.pharmgkb.org/pathway/PA145011108