Farmakoterapie cystické fibrózy
Cystická fibróza (CF) je vrozené onemocnění vyvolané mutacemi genu pro transmembránový regulátor vodivosti (CFTR) s plicními a mimoplicními projevy. Farmakoterapie plicního postižení u CF je cílena především na mukostázu, bakteriální infekci a neutrofilní zánět. Léčba mimoplicních projevů CF se týká zejména pankreatické insuficience, deficitu vitaminů rozpustných v tucích, střevní obstrukce, diabetu, hepatopatie a osteoporózy. Ve fázi výzkumu je v současnosti genová léčba CF a terapie cílená na jednotlivé mutace genu CFTR.
Úvod
Cystická fibróza (CF) je vrozené onemocnění vyvolané mutacemi genu pro transmembránový regulátor vodivosti (CFTR). Dědičnost tohoto onemocnění je autozomálně recesivní. V České republice činí incidence přibližně 1 : 2700 živě narozených a v roce 2007 bylo sledováno 471 pacientů s mediánem věku 14,5 roku (starších 18 let bylo 38,2 %).
Mutace genu CFTR v současnosti dělíme podle patogeneze do tříd I až V, přičemž dle klinických projevů patří mutace třídy I až III mezi tzv. těžké a třídy IV a V mezi tzv. mírné. Např. celosvětově nejčastější mutace F508del je řazena do třídy II, charakterizované poruchami nitrobuněčného transportu a vyzrávání CFTR proteinu. CFTR protein je cAMP regulovaný chloridový kanál, jehož dysfunkce vede k postižení žláz se zevní sekrecí. Kromě chloridových iontů se CFTR protein podílí na transportu dalších aniontů (thiokyanáty, bikarbonáty) a nízkomolekulárních látek (glutathion) a na regulaci dalších iontových kanálů, především epiteliálního natriového kanálu (ENaC), jehož aktivita je CFTR proteinem tlumena. U CF tak dochází vlivem dysfunkce CFTR proteinu ke zvýšení aktivity ENaC a k hyperabsorpci natriových iontů (a sekundárně i vody), což vede k zahuštění hlenu na sliznicích dýchacího a trávicího ústrojí [1]. Potní žlázy nemocných CF produkují pot obsahující zvýšenou koncentraci NaCl. U nemocných tak může dojít k tzv. syndromu ztráty solí, vedoucímu akutně k hypotonické dehydrataci a šokovému stavu, chronicky pak k metabolické alkalóze. Vyšetření chloridů v potu je (spolu s vyšetřením mutací genu CFTR) využíváno i diagnosticky, patologické jsou hodnoty > 60 mmol/l, hraniční pak 30–60 mmol/l.
V dýchacích cestách jedinců s CF dochází k mukostáze, chronické bakteriální infekci (typické patogeny představují Staphylococcus aureus, Haemophilus influenzae, Pseudomonas aeruginosa a komplex Burkholderia cepacia) a k chronickému neutrofilnímu zánětu. Proteázy a volné kyslíkové radikály uvolňované z leukocytů poškozují dýchací cesty a vedou k rozvoji bronchiektazií s obstrukční ventilační poruchou. Plicní onemocnění má období stability a exacerbací (obvykle při vzplanutí respirační infekce) a je zodpovědné za úmrtí 95 % nemocných CF. Dále se v oblasti dýchacího ústrojí můžeme u CF setkat se sinusitidami, nosní polypózou, atelektázami, pneumothoraxy (PNO), hemoptýzami, CF-astmatem, alergickou bronchopulmonální aspergilózou (ABPA) a netuberkulózními mykobakteriózami (NTM).
V trávicím ústrojí vede postižení pankreatu k fibróze a cystické přestavbě a při rozsáhlejší destrukci i k diabetu mellitu (CFRD). Insuficience zevní sekrece pankreatu se vyskytuje u 85 % případů CF, má za následek steatoreu a malnutrici a podílí se na rozvoji osteoporózy (CFRBD). Během života se může rozvinout i u původně pankreaticky suficientních osob (ty jsou rovněž ohroženy rizikem recidivujících pankreatitid). Kromě malnutrice dochází i k malabsorpci vitaminů rozpustných v tucích (A, D, E a K). Hepatobiliární postižení vede především k fibróze až cirhóze jater (CFLD) a cholelitiáze. Střevní postižení ohrožuje adolescenty a dospělé pacienty rozvojem ileózního stavu při syndromu obstrukce distálního střeva (DIOS), jenž je ekvivalentem mekoniového ileu novorozenců. U části nemocných je přítomen gastroezofageální reflux (GER).
Obstruktivní azoospermie je způsobena kongenitální bilaterální absencí vas deferens (CBAVD) a nacházíme ji u 98 % mužů s CF jako nejkonstantnější projev této choroby. Uvedené klinické projevy CF mohou být různě vyjádřeny s ohledem na typ mutací genu CFTR (mírnými mutacemi se vyznačují variabilní sinopulmonální onemocnění, bývá přítomna pankreatická suficience a pankreatitidy, nikoli však CFRD a CFLD). Roli hrají rovněž modifikující geny, například TGF-b u plicního nebo a1-antitrypsin u hepatálního postižení. Z faktorů zevního prostředí je důležitý druh chronické bakteriální infekce dýchacích cest (prevence infekce patří mezi zcela zásadní režimová opatření u CF!) a dostupnost moderních léčebných metod [2].
Léčba cystické fibrózy
Léčba CF je velice komplexní záležitostí, která zahrnuje farmakologické i nefarmakologické postupy [3].
Nefarmakologické léčebné metody
V oblasti výživy je pro nemocné CF zcela zásadní vysokokalorická dieta. Ve srovnání s normou potřebují tito pacienti o 20–50 % vyšší kalorický příjem, ve kterém tuky představují 35–45 % (podmínkou je adekvátní substituce pankreatickými enzymy u pankreaticky insuficientních jedinců). U nemocných, kteří nedosahují adekvátního energetického příjmu stravou, ordinujeme nutriční podporu (sipping, sondová nebo parenterální výživa). Vážnou chybou je ordinace diabetické diety nemocným s CFRD, neboť tato je pro ně vysloveně karenční (ve stravě omezujeme pouze volné cukry, které nahrazujeme tuky). V péči o dýchací ústrojí má nezastupitelnou roli respirační fyzioterapie. Ta využívá aktivní cyklus dechových technik, autogenní drenáž a rovněž instrumentální techniky, především přístroje vytvářející kontinuální či oscilující pozitivní tlak v exspiriu (PEP maska, flutter a další). Nemocní jsou zacvičeni v provádění těchto dechových cvičení s cílem evakuovat co nejvíce sekretů z dýchacích cest s co nejmenším úsilím, a to obvykle 2krát denně (v období plicních exacerbací častěji).
Dalším důležitým postupem v léčbě bronchopulmonálního onemocnění je inhalační léčba využívaná k podávání aerosolových nebo práškových forem léčebných přípravků. Kromě běžných dávkovaných aerosolů (MDI) a práškových inhalátorů (DPI) používáme k podávání roztoků léčiv výkonné kompresorové inhalátory s tryskovými nebulizátory. Novinkou je elektronický inhalátor pracující na principu oscilující membrány (PARI eFlow Rapid®), který výrazně zkracuje dobu inhalace. Nutné je i zde zdůraznit adekvátní péči o inhalační pomůcky s cílem zabránit infekci dýchacích cest.
Oxygenoterapie a mechanická ventilace (preferována je neinvazivní cesta) se týká nemocných s respirační insuficiencí při těžkých plicních exacerbacích nebo při pokročilém plicním onemocnění. Chirurgická, endoskopická a intervenční radiologická léčba se u CF podílí na řešení problémů souvisejících se sinusitidami, nosní polypózou, atelektázami, PNO, hemoptýzami, GER, portální hypertenzí, cholelitiázou a ileózními stavy. Zvláštní kapitolu představují transplantace plic a jater v případě selhávání těchto orgánů a metody asistované reprodukce u mužů.
V neposlední řadě je třeba zmínit i psychoterapeutickou podporu poskytovanou nemocným a jejich blízkým, jejíž potřeba vyplývá z povahy onemocnění.
Farmakoterapie plicního onemocnění u CF
Farmakologické možnosti ovlivnění bronchopulmonálního onemocnění u CF zahrnují s ohledem na patogenezi choroby především mukoaktivní, antimikrobiální a protizánětlivé léky [4]. Farmakologická léčba dalších projevů postižení dýchacího ústrojí u CF (sinusitidy, nosní polypóza, atelektázy, PNO, hemoptýzy, CF-astma, ABPA a NTM) se neliší od postupů užívaných u jiných chorob a její výčet přesahuje rámec tohoto sdělení.
Mukoaktivní léky u CF
K farmakologickému ovlivnění porušené mukociliární clearance u CF je k dispozici řada preparátů, viz obr. 1. Jde jednak o všeobecně používané léky, jako je ambroxol, acetylcystein a mesna, jednak o léky pro CF specifické, a to milimolární roztok amiloridu, hypertonický roztok NaCl a alfadornáza. Řada dalších léků specifických pro CF je pak v současnosti předmětem výzkumu a klinických studií [5].
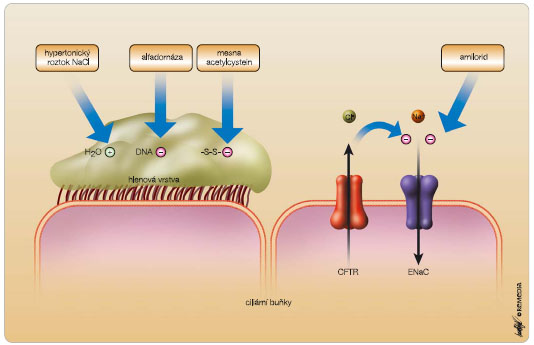 Amilorid je blokátorem ENaC a jeho podání snižuje absorpci Na+ tímto kanálem. Dochází tak ke zlepšení hydratace sekretů dýchacích cest a jejich snazší expektoraci. Užití amiloridu je specialitou českých zemí, jinde ve světě se prakticky neužívá. Jeho nevýhodou je krátký biologický poločas (23 minut) na sliznici dýchacích cest a účinnost spíše u lehčích stadií plicního postižení. V praxi využíváme milimolární roztok připravovaný jako magistraliter (Rp. Amiloridi chlorati 0,06, Natrii chlorati 1,80, Aquae destilatae ad 200,0, D.S. 2 x 3 ml k inhalaci).
Amilorid je blokátorem ENaC a jeho podání snižuje absorpci Na+ tímto kanálem. Dochází tak ke zlepšení hydratace sekretů dýchacích cest a jejich snazší expektoraci. Užití amiloridu je specialitou českých zemí, jinde ve světě se prakticky neužívá. Jeho nevýhodou je krátký biologický poločas (23 minut) na sliznici dýchacích cest a účinnost spíše u lehčích stadií plicního postižení. V praxi využíváme milimolární roztok připravovaný jako magistraliter (Rp. Amiloridi chlorati 0,06, Natrii chlorati 1,80, Aquae destilatae ad 200,0, D.S. 2 x 3 ml k inhalaci).
Hypertonický roztok NaCl (hypertonic saline, HS) vede rovněž ke zlepšení hydratace sekretů dýchacích cest, a to osmotickým účinkem: Zlepšení mukociliární clearance po podání HS přetrvává více než 8 hodin. Využití HS je záležitostí posledních let, neboť teprve v roce 2006 byly publikovány dvě studie [6, 7], které jeho podávání doslova vydláždily cestu. V praxi lze použít 3–7% HS (v uvedených studiích byl použit 7% HS). Sami využíváme molární roztok (5,85%), připravovaný opět jako magistraliter (Rp. Natrii chlorati 16,38, Aquae destilatae ad 280,0, D.S. 2 x 4 ml k inhalaci). Před podáním HS se doporučuje premedikace inhalačními bronchodilatancii (např. 0,1 mg salbutamolu ve formě MDI).
Alfadornáza neboli rekombinantní humánní deoxyribonukleáza využívá ke zlepšení mukociliární clearance jiný mechanismus, a to snížení vizkozity sekretů dýchacích cest degradací molekul DNA z rozpadlých bakterií, epitelií a leukocytů. V USA je doporučena pro léčbu všech nemocných s CF, reálně je však podávána u 76,1 % nemocných. V ČR ji lze předepsat při nedostatečné účinnosti inhalačních mukolytik nebo amiloridu a při častých respiračních infekcích vyžadujících antibiotickou léčbu. V populaci dospělých s CF je v ČR využívána u 75,7 % pacientů. Alfadornázu podáváme v dávce 2,5 mg 1krát denně (při výrazném zahlenění a v období plicních exacerbací lze podávat 2krát denně) obvykle v odpoledních hodinách. K podání nesmí být používány ultrazvukové nebulizátory z důvodu degradace molekul alfadornázy. Pokud po inhalaci následuje respirační fyzioterapie, je třeba dodržet odstup nejméně jedné hodiny (lépe dvou) tak, aby byl dostatek času na působení enzymu na DNA. Základní nevýhodou alfadornázy je její vysoká cena [8]. Z řady mukoaktivních léků testovaných pro využití u CF je třeba zmínit především mannitol podávaný v práškové inhalační formě (zkrácení doby inhalací) jako osmoticky působící agens a dále denufosol a duramycin (aktivátory alternativních chloridových kanálů) podávané inhalačně v roztoku.
Antimikrobiální léčba u CF
Antimikrobiální farmakoterapie je u CF užívána z řady důvodů. Kromě terapie plicních exacerbací jde rovněž o léčbu preventivní, eradikační a chronickou supresivní. U CF se podávají antibiotika ve vyšších dávkách a po delší dobu, než je běžné. Využívána je i inhalační antibiotická léčba a domácí intravenózní antibiotická léčba [9]. U aminoglykosidů preferujeme podávání jedenkrát denně za kontrol sérových hladin, zkouší se i kontinuální podávání betalaktamových antibiotik.
Preventivní léčba je v některých centrech podávána u dětí v prvních dvou letech života k prevenci infekce S. aureus (SA). Užívány bývají dlouhodobě podávané protistafylokokové betalaktamy.
Eradikační léčba je indikována při každém novém záchytu patogenu. Cílem je zabránit chronické kolonizaci dýchacích cest a častým plicním exacerbacím. Podmínkou jsou pravidelné odběry respiračních sekretů, ideálně každý měsíc, minimálně však čtvrtletně. Volba preparátu se řídí zjištěnou citlivostí patogenu, často jsou podávány i kombinace antibiotik. Doba podávání činí nejméně dva týdny. Eradikační léčba je zcela zásadním opatřením při záchytu P. aeruginosa (PA). Tuto bakterii nacházíme v respiračních sekretech až u 80 % dospělých s CF a chronická infekce tímto patogenem zkracuje přežití přibližně o 10 let. Klasické eradikační schéma zahrnuje podání ciprofloxacinu perorálně po dobu 3 týdnů a kolistinu inhalačně po dobu 3 měsíců. Nově jsou dosahovány dobré výsledky i s vysokodávkovaným inhalačním tobramycinem podávaným po dobu 28 dnů.
Chronická supresivní léčba se týká především PA, a to v případě chronické infekce dýchacích cest spojené se zrychleným poklesem plicních funkcí, zhoršováním stavu výživy a projevy systémové zánětlivé reakce (vzestup hladiny CRP a leukocytů). Zde využíváme dlouhodobé inhalační podávání kolistinu (kontinuálně 1–2 MIU 2krát denně) nebo vysokodávkovaného tobramycinu (cyklicky po 28 dnech 300 mg 2krát denně). Supresivní léčbu nepoužíváme při chronické infekci komplexem B. cepacia (BC).
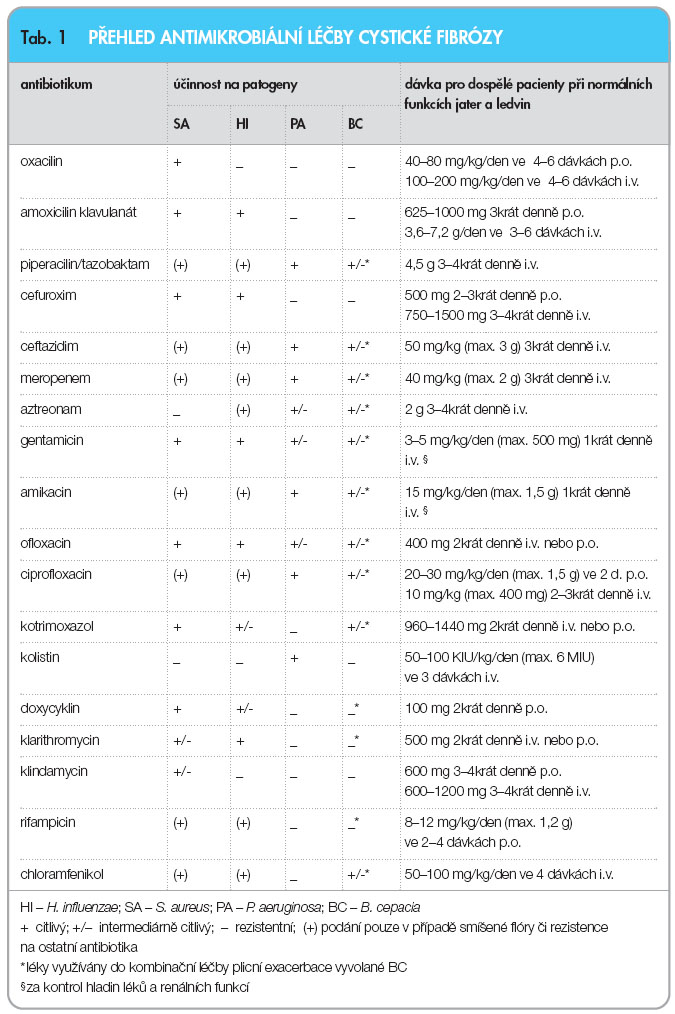
Při léčbě plicní exacerbace se volba preparátu, cesta podání i doba léčby řídí obdobnými zásadami jako léčba eradikační. V případě záchytu více patogenů nebo rezistentních kmenů PA a BC podáváme běžně kombinace antibiotik zahrnující 1–2 betalaktamy a/nebo 1–3 antibiotika dalších tříd. Během léčby plicní exacerbace je nutné monitorovat klinickou odpověď, za hospitalizace alespoň jedenkrát týdně kontrolujeme zánětlivé parametry, plicní funkce i mikrobiologický nález v respiračních sekretech. V případě neúspěchu indikujeme změnu antimikrobiální terapie. Přehled antibiotik běžně používaných u CF včetně dávkování pro dospělé a účinnosti na obvyklé patogeny je uveden v tab. 1. Meticilin rezistentní SA zatím nepředstavuje u nemocných CF v ČR takový problém jako v zahraničí (např. v USA je přítomen u 22,6 % pacientů). V případě jeho záchytu je lékem první volby kotrimoxazol, v případě rezistence lze užít vankomycin (i inhalačně), linezolid či tigecyklin. U mykotických agens (Candida albicans, Aspergillus fumigatus) užíváme azolová antimykotika nebo preparáty amfotericinu B (i inhalačně) v obvyklých dávkách. Z virologické problematiky je u CF doporučena každoroční vakcinace proti viru influenzy A.
 Klinický výzkum v oblasti antimikrobiální léčby se týká hlavně inhalačních forem antibiotik. Kromě již dlouhodobě užívaného kolistinu a vysokodávkovaného tobramycinu byl v letošním roce v USA schválen ke klinickému použití aztreonam jako další protipseudomonádové antibiotikum (v ČR zatím není dostupný). V současnosti probíhají klinické studie především s lipozomální formou amikacinu k inhalaci a s práškovými inhalačními formami kolistinu a tobramycinu.
Klinický výzkum v oblasti antimikrobiální léčby se týká hlavně inhalačních forem antibiotik. Kromě již dlouhodobě užívaného kolistinu a vysokodávkovaného tobramycinu byl v letošním roce v USA schválen ke klinickému použití aztreonam jako další protipseudomonádové antibiotikum (v ČR zatím není dostupný). V současnosti probíhají klinické studie především s lipozomální formou amikacinu k inhalaci a s práškovými inhalačními formami kolistinu a tobramycinu.
Protizánětlivá léčba u CF
Chronický neutrofilní zánět hraje zásadní roli v patogenezi plicního postižení u CF. Snaha o jeho potlačení je tedy logickým krokem v léčbě těchto nemocných. Klinického využití, byť často kontroverzního, zatím dosáhly kortikosteroidy, ibuprofen a azithromycin. Systémové kortikosteroidy nejsou s výjimkou terapie ABPA k rutinní léčbě zánětu u CF doporučeny. Podávání prednisonu v dávce 1–2 mg/kg obden sice zpomaluje progresi nemoci, ale nežádoucí účinky jsou považovány za nepřijatelné [3]. V současnosti se nicméně připouští krátkodobé podávání kortikosteroidů v případě těžkých plicních exacerbací. Sami rovněž podáváme na individuální bázi nízké dávky prednisonu (5–20 mg obden) u nemocných s těžkou ventilační poruchou a chronickou infekcí dýchacích cest, kterou nelze plně kontrolovat antibiotiky. Efekt léčby hodnotíme po 3–6 měsících a poté se rozhodujeme, zda léčbu ukončit, či v ní pokračovat. Obdobný přístup používáme i v případě inhalačních kortikosteroidů. Ty jsou indikovány u CF-astmatu (občas i s prokazatelným zvýšením množství vydechovaného NO, který je jinak u CF typicky snížen), kde podáváme beklometason, budesonid či flutikason v obvyklých dávkách. Výsledky studií s léčbou inhalačními kortikosteroidy u nemocných bez CF-astmatu jsou rozporné. Jejich klinické využití je v řadě center sice časté, ale také často kritizované. Důvodem jsou výsledky studie CF WISE [10], jež svědčily pro to, že u většiny nemocných s CF lze podávání těchto léků bezpečně ukončit.
Dalším protizánětlivým lékem u CF je vysokodávkovaný ibuprofen. Jeho podávání je dle americké CF Foundation doporučeno u pacientů ve věku 6–12 let s FEV1 > 60 % náležitých hodnot. Podává se v dávce 60 mg/kg/den rozdělených do dvou dílčích, nutné je monitorování sérových hladin (50–100 mg/l). V našich podmínkách nejde o běžnou léčbu a rovněž v USA je podáván pouze u 3,8 % pacientů podle výše uvedených kritérií. Nezanedbatelným faktorem je zde nepochybně otázka gastrointestinální tolerance. U nemocných s chronickou infekcí PA ve věku ≥ 6 let je vhodným lékem nízkodávkovaný azithromycin. Přesný mechanismus jeho protizánětlivého účinku dosud není znám. Podle studií publikovaných v letech 2002–2003 vede tato léčba ke zlepšení plicních funkcí, poklesu lokálních i systémových markerů zánětu a ke snížení výskytu plicních exacerbací [11]. Azithromycin podáváme 3krát týdně (režim pondělí–středa–pátek) v dávce 500 mg p.o. u osob s tělesnou hmotností vyšší než 40 kg (resp. 250 mg v případě tělesné hmotnosti 25–40 kg). Režim podávání dávky 500 mg 3krát týdně lze modifikovat na 250 mg denně. Rovněž zde je doporučen individuální přístup se zhodnocením efektu léčby po 6 měsících a s rozhodnutím o jejím pokračování či ukončení. Během terapie je třeba pravidelně kontrolovat jaterní testy a přítomnost netuberkulózních mykobakterií ve sputu. Výzkum v oblasti protizánětlivé léčby je bohatý. Je možné uvést např. antiproteázy a glutathion (obojí k inhalačnímu podávání), n-3 polynenasycené mastné kyseliny, sildenafil, imunosupresiva (ciklosporin a methotrexát) a imunoglobuliny. U nemocných s ABPA se zkouší léčba monoklonální anti-IgE protilátkou (omalizumab). Žádný z těchto přístupů však zatím nedosáhl klinického využití.
Farmakoterapie mimoplicních projevů CF
Podrobný rozbor farmakoterapie mimoplicního postižení u CF přesahuje rámec tohoto sdělení. U řady projevů se léčba navíc neliší od běžné populace (např. GER, pankreatitida a další). Proto bude v této části stručně zmíněna léčba pankreatické insuficience, malabsorpce vitaminů rozpustných v tucích a DIOS, dále léčba tzv. velkých komplikací dospělého věku, tedy CFRD, CFLD a CFRBD.
Základním způsobem léčby pankreatické insuficience je substituce pankreatických enzymů. Je nutné používat moderní preparáty ve formě acidorezistentních mikrotablet v želatinových kapslích. Pouze tímto způsobem substituce lze současně využívat vysokokalorickou dietu bohatou na tuky. Pankreatickou insuficienci verifikujeme pomocí vyšetření elastázy 1 ve stolici (patologické jsou hodnoty < 100 mg/g). U kojenců se zahajuje dávkou 400–800 IU lipázy na 1 g tuku v potravě, u větších dětí a dospělých dávkou 500 IU lipázy/kg ke svačině a 1000 UI lipázy/kg k hlavním jídlům. Dávka substituce se pak titruje tak, aby měl nemocný maximálně tři neprůjmové stolice denně. Celková denní dávka nemá přesáhnout 10 000 UI lipázy/kg vzhledem k riziku rozvoje fibrotizující kolonopatie. Není-li tato dávka dostatečná, doporučuje se přidat inhibitory protonové pumpy nebo blokátory H2 ke snížení žaludeční acidity. Nutno upozornit na skutečnost, že pankreatickou substituci musí tito nemocní dostávat i v případě plné parenterální výživy, a to vzhledem k riziku obstrukce střev nestráveným hlenem. V těchto případech podáváme 10 000 UI lipázy každé čtyři hodiny [3].
Suplementace vitaminů rozpustných v tucích je s výjimkou vitaminu K nutná u většiny nemocných s pankreatickou insuficiencí. Dávky vitaminu A, D a E upravujeme podle sérových hladin (hladinu vitaminu D kontrolujeme v podzimních měsících). Obvyklé denní dávky pro dospělé jsou 10 000–20 000 IU vitaminu A, 400–800 IU vitaminu D a 200–600 mg vitaminu E. Vitamin K doplňujeme u nemocných s jaterní cirhózou, osteoporózou, těžkými formami pankreatické insuficience a při opakované antibiotické léčbě. Obvyklá dávka pro dospělé je 10 mg fytomenadionu denně. Z dalších nutrientů je třeba upozornit na zvýšenou potřebu suplementace NaCl v období horka, protrahovaných horeček nebo při těžké fyzické práci. U části nemocných je rovněž nutné doplňovat preparáty železa v závislosti na sérových hladinách. Zde musíme brát na zřetel interakce s antacidy a pankreatickými enzymy, které absorpci železa snižují [3].
DIOS (Distal Intestinal Obstruction Syndrome) je komplikace s variabilním klinickým obrazem, kdy dochází k různě těžké poruše pasáže v oblasti ileocékálního přechodu při přítomnosti zahuštěného hlenu. Nutná je adekvátní hydratace, z farmakoterapeutických opatření přichází v úvahu zvýšení dávky pankreatické substituce a podání mukolytik (acetylcystein) u lehčích forem a osmoticky působících laxativ (makrogoly) u těžších forem DIOS (nejde-li již o ileus, kde je nutný chirurgický výkon) [3].
CFRD (Cystic Fibrosis-Related Diabetes) se typicky rozvíjí ve věku 18–21 let a je přítomen asi u třetiny dospělých s CF. Kromě výše uvedeného zákazu diabetické diety nepoužíváme v léčbě perorální antidiabetika, ale výhradně inzulin. Začínáme dlouhodobě působícími preparáty v nízké dávce jednou denně a s klesající inzulinovou produkcí dávky zvyšujeme, přecházíme na směsné inzuliny podávané dvakrát denně a posléze i na intenzifikované režimy. Důvodem k zahájení léčby inzulinem nemusí být jen prokázaný diabetes. V případě těžké inzulinopenie (zjišťované intravenózním glukózovým tolerančním testem) zahajujeme léčbu i tehdy, pokud se bez jiné zjistitelné příčiny zhoršuje stav výživy a plicních funkcí [12].
CFLD (Cystic Fibrosis Liver Disease) se typicky rozvíjí u dětí ve věku 5–10 let a v dospělosti se pak setkáváme s pokročilejšími formami této komplikace. Fokální nebo multilobulární biliární fibróza až cirhóza jater se nachází u 20–40 % nemocných CF a je zodpovědná za 2–5 % případů úmrtí u této choroby. Léčebně zasahujeme podáváním kyseliny ursodeoxycholové v dávce 20 mg/kg/den rozdělené do dvou dílčích. Při této léčbě je popisováno zlepšení výsledků jaterních biochemických testů a histologického nálezu a rovněž stavu výživy a metabolismu esenciálních mastných kyselin. Léčba projevů portální hypertenze se neliší od jaterních cirhóz jiné etiologie [13].
CFRBD (Cystic Fibrosis-Related Bone Disease) se v různě těžké formě nachází u 50–75 % dospělých s CF. V patogenezi hraje roli celá řada faktorů, především malnutrice a systémová zánětlivá reakce na plicní infekt. V léčbě kromě suplementace kalcia a vitaminu D využíváme především antiresorpční terapii bisfosfonáty v obvyklých dávkách. U nemocných s opožděnou pubertou a projevy hypogonadismu přichází v úvahu i hormonální léčba [14].
Farmakoterapie cílená na „basic defect“ u CF
V této oblasti se výzkum ubírá především dvěma směry. Jednak je to genová léčba, jednak farmaka cílená na jednotlivé mutace genu CFTR [15]. Ačkoli od objevu genu CFTR uběhlo letos již 21 let, je genová léčba pro nemocné CF nedostupná a předpokládá se, že tento stav potrvá ještě nejméně pět, ale spíše deset let. V současnosti se jedná o experimentální terapie využívající jednak virové (např. adenoasociované viry) a jednak nevirové vektory (např. na bázi kationických lipidů). Kromě genové léčby je předmětem intenzivního výzkumu i tzv. CFTR-farmakoterapie. Jde o vývoj látek cílených na jednotlivé mutace genu CFTR. Ve třídě I (tzv. stop mutace) jde o látky umožňující přemostění předčasných stop kodonů. Mezi ně patří např. aminoglykosidová antibiotika a ataluren. Ve třídě II charakterizované poruchami nitrobuněčného zpracování a transportu CFTR proteinu je nadějným lékem korektor VX-809 určený pro celosvětově nejčastější mutaci F508del. Ve třídě III spojené s poruchami aktivace CFTR proteinu v buněčné membráně je to pak potenciátor VX-770 cílený na mutaci G551D (tzv. keltská mutace, hojná i v ČR), který je v současné době ve III. fázi klinického hodnocení léčiv (probíhá mj. i na našem pracovišti).
Závěr
Seznam použité literatury
- [1] Bush A, Alton EW, Davies JC, et al. Cystic fibrosis in the 21st century. Karger, Basel 2006, 329 s.
- [2] Hodson M, Geddes D, Bush A, et al. Cystic fibrosis 3rd ed. Hodder Arnold, London 2007, 503 s.
- [3] Vávrová V, a kol. Cystická fibróza. Grada Publishing, Praha 2006, 516 s.
- [4] Flume PA, O'Sullivan BP, Robinson KA, et al. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med 2007; 176: 957–969.
- [5] Rubin BK. Mucus, phlegm, and sputum in cystic fibrosis. Respir Care 2009; 54: 726–732.
- [6] Donaldson SH, Bennett WD, Zeman KL, et al. Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N Engl J Med 2006; 354: 241–250.
- [7] Elkins MR, Robinson M, Rose BR, et al. National Hypertonic Saline in Cystic Fibrosis (NHSCF) Study Group. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med 2006; 354: 229–240.
- [8] Fuchs HJ, Borowitz DS, Christiansen DH, et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Engl J Med 1994; 331: 637–642.
- [9] Cystic Fibrosis Trust. Antibiotic treatment in cystic fibrosis. 2009, 102 s. Dostupné na: http://www. cftrust.org.uk/aboutcf/publications/consensusdoc/Antibiotic_treatment_for_Cystic_Fibrosis.pdf.
- [10] Balfour-Lynn IM, Lees B, Hall P, et al. Multicenter randomized controlled trial of withdrawal of inhaled corticosteroids in cystic fibrosis. Am J Respir Crit Care Med 2006; 173: 1356–1362.
- [11] Saiman L, Marshall BC, Mayer-Hamblett N, et al. Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa: a randomized controlled trial. JAMA 2003; 290: 1749–1756.
- [12] Moran A, Hardin D, Rodman D, et al. Diagnosis, screening and management of cystic fibrosis related diabetes mellitus. A consensus conference report. Diabetes Res Clin Pract 1999; 45: 61–73.
- [13] Colombo C, Russo MC, Zazzeron L, et. al. Liver disease in cystic fibrosis. J Pediatr Gastroenterol Nutr 2006; 43: S49–S55.
- [14] Aris RM, Merkel PA, Bachrach LK, et al. Consensus statement: Guide to bone health and disease in cystic fibrosis. J Clin Endocrinol Metab 2004; 90: 1888–1896.
- [15] Ratjen FA. Cystic fibrosis: Pathogenesis and future treatment strategies. Respir Care 2009; 54: 595–602.