Intravenózní imunoglobulin u autoimunitních nervosvalových onemocnění
Intravenózní imunoglobulin (IVIG) je často indikován u různých neurologických onemocnění. Nejčastější využití je u autoimunitních nervosvalových chorob. Jeho mechanismus účinku je komplexní s ovlivněním humorální i buněčné imunity. Kontrolované klinické studie prokazují efekt IVIG u většiny autoimunitních nervosvalových chorob. U Guillainova-Barréova syndromu je IVIG a léčebná plazmaferéza léčbou první volby a mají stejnou účinnost (síla doporučení A). Léčbu IVIG provází méně vedlejších účinků, a proto se mu dává přednost (síla doporučení B). U chronické zánětlivé demyelinizační polyneuropatie se středně těžkou až těžkou disabilitou je vhodné použít jako léčbu 1. volby IVIG nebo kortikoidy (síla doporučení A). Vzhledem k chybění alternativní léčby je IVIG u multifokální motorické neuropatie lékem první volby (síla doporučení A). U myasthenia gravis je léčba exacerbace léčebnou plazmaferézou i IVIG stejně účinná (síla doporučení A). Podávání IVIG se doporučuje u dermatomyozitidy jako léčba třetí linie v kombinaci s prednisonem, a to u pacientů, u kterých se po adekvátní terapii kortikoidy a jejich kombinací s imunosupresivy neprojevil dostatečný efekt léčby (síla doporučení B). IVIG má příznivý léčebný profil. Jeho vysoká cena je kompenzována nižšími náklady na intenzivní a následnou péči a zvýšenou kvalitou života.
Úvod
Autoimunitní nervosvalová onemocnění jsou heterogenní skupinou chorob, jejichž společným jmenovatelem je poškození periferních nervů, nervosvalové ploténky nebo příčně pruhovaných svalů autoimunitním procesem. Mezi zánětlivé polyneuropatie patří Guillainův-Barréův syndrom (GBS) a jeho varianty, chronická zánětlivá demyelinizační polyneuropatie (CIDP), multifokální motorická polyneuropatie (MMN) a některé vzácné polyneuropatie paraneoplastické či asociované s paraproteinemií. Mezi nejčastější onemocnění nervosvalového přenosu patří myasthenia gravis (MG), mnohem vzácnější je Lambertův-Eatonův syndrom (LEMS). Zánětlivé myopatie jsou zastoupeny dermatomyozitidou (DM), polymyozitidou (PM), autoimunitní nekrotizující myozitidou a myozitidou s inkluzními tělísky. Prevalence těchto onemocnění je minimálně 30/100 000, přičemž přímé náklady vynaložené na léčbu, případně na sociální dávky ve spojitosti s trvalou invaliditou, jsou poměrně velké [1–4]. Intravenózní imunoglobulin (IVIG) představuje významný pokrok v léčbě autoimunitních onemocnění. Přesto, že je cena IVIG vysoká (1200–1400 Kč za 1 gram), představuje jeho zavedení do léčebné praxe redukci celkových vynaložených prostředků na léčbu. Např. u pacientů s GBS nebo u pacientů s myastenickou krizí IVIG významně zkracuje dobu nutné mechanické plicní ventilace a celkovou dobu hospitalizace, a to při relativně velmi dobrém bezpečnostním profilu [5]. Cílem práce je podat přehled o možnostech léčby autoimunitních nervosvalových onemocnění IVIG se zaměřením na výsledky kontrolovaných klinických studií a doporučení pro praxi.
Imunopatogeneze autoimunitních nervosvalových onemocnění
Autoimunitní nervosvalové choroby jsou důsledkem ztráty imunitní tolerance k autoantigenům na základě defektní klonální delece a dalších mechanismů, např. molekulárních mimikry. Antigenní epitopy mikrobiálních agens jsou prostřednictvím antigen prezentujících buněk rozpoznány MHC molekulami II. třídy jako tělu vlastní a tím vedou za přispění kostimulačních molekul ke klonální expanzi T buněk. Tyto buňky pak prostupují hematoneurální bariérou do cílových tkání, kde vykazují cytotoxický efekt [6–8]. Autoprotilátky produkované B buňkami v interakci s T buňkami, které produkují interleukin 4 a 6, rozpoznávají cílové neurální nebo svalové proteiny prostřednictvím makrofágů nebo komplementu. Cytokiny produkované T buňkami zvyšují aktivitu intercelulárních a vaskulárních adhezních molekul nebo matrixové metaloproteinázy v endoteliálních buňkách a umožňují zvýšenou propustnost aktivovaných lymfocytů, cytokinů a stimulovaných makrofágů cévní stěnou vazbou na Fc receptory. Dochází k dysregulaci prostřednictvím dysfunkce regulujících T lymfocytů se zvýšenou produkcí prozánětlivých cytokinů. U GBS je imunopatogeneze spojena s produkcí protilátek reagujících s bakteriálními komponentami, zejména s Campylobacter jejuni, které napodobují neurální glykolipidy a gangliosidy, čímž je prolomena imunitní tolerance. U pacientů s patrnou axonální degenerací byla prokázána imunopatogenní role IgG protilátek proti GM1, GD1b nebo GD1a gangliosidům, které jsou exprimovány na periferních nervech [9–12]. Protilátky proti GQ1b gangliosidům jsou úzce asociovány s Millerovým-Fisherovým syndromem [13]. U CIDP byly stejně jako u GBS prokázány molekulární mimikry, antiglykolipidové protilátky a dysregulace T buněk [14, 15]. Autoimunitní podklad MMN podporuje kromě terapeutické odezvy řada biochemických nálezů, zejména obvyklý nález vysokých titrů autoprotilátek proti GM1 gangliosidům. Anti-glykolipidové protilátky nemají zřejmě patogenní význam, jsou pouze markery onemocnění. Zajímavou a nedořešenou otázkou zůstává, proč jsou postiženy selektivně motorické nervy. Demyelinizace nejspíše postihuje pouze motorické fascikuly nervu; potenciální cílový antigen může být odlišný pro myelin motorických a senzitivních nervových vláken [3, 16, 17]. U zánětlivých polyneuropatií mohou aktivované makrofágy napadnout myelin nebo podporovat produkci zánětlivých cytokinů, nehledě na roli autoprotilátek, které aktivují komplement a membrány atakující komplex (MAC). Tyto protilátky mohou poškozovat myelin též přímou vazbou na Fc receptory aktivovaných makrofágů [18, 19]. Podobné imunitně zprostředkované mechanismy hrají primární roli v imunopatogenezi zánětlivých myopatií. Sérum pacientů s aktivní DM obsahuje vysoké hladiny komplementu a MAC, které jsou uloženy ve formě depozit v oblasti kapilár, které podléhají destrukci. U PM dochází ke klonální expanzi CD8+ cytotoxických buněk, které vedou k endomysiální infiltraci a invazi do svalových vláken exprimujících MHC molekuly I. třídy. Nekrotizující autoimunitní myozitida je charakterizována poškozením svalových vláken prostřednictvím makrofágů. U myozitidy s inkluzními tělísky panují pochybnosti o autoimunitní etiologii onemocnění [20–22]. MG je prototypem imunitně zprostředkovaného onemocnění nervosvalového přenosu. Byly rozpoznány dva cílové antigeny, nikotinový acetylcholinový receptor a svalově specifická tyrozinkináza, vůči nimž jsou namířeny IgG protilátky, které spolu s komplementem a MAC blokují nebo strukturálně poškozují acetylcholinové receptory na postsynaptické membráně nervosvalové ploténky a tím omezují nervosvalový přenos [23]. U LEMS jsou autoprotilátky namířeny proti vysokonapěťovým kalciovým kanálům typu P/Q na terminálním zakončení motorických nervů. U paraneoplastické formy LEMS se uplatňují molekulární mimikry, kdy autoprotilátky zkříženě reagují s antigenními epitopy malobuněčného plicního karcinomu [24].
Imunomodulační mechanismy intravenózního imunoglobulinu
![Obr. 1 Imunomodulační mechanismy IVIG u autoimunitních nervosvalových chorob; volně podle [25–27] – Dalakas, 1999; Bayry, et al., 2003; Quick, et al., 2011. Intravenózní imunoglobulin (IVIG) moduluje řadu imunologických pochodů (A–F), které se podílejí na imunopatogenezi autoimunitních nervosvalových onemocnění (blíže v textu). Jsou uvedeny specifické léčebné účinky na základě experimentálních důkazů. Jinými předpokládanými mechanismy IVIG, které nejsou uvedeny, jsou např. narůstající katabolismus IgG, alterace efektorových funkcí T buněk a modulace apoptózy. CIDP – chronická zánětlivá demyelinizační polyneuropatie; DM – dermatomyozitida; GBS – Guillainův-Barréův syndrom; ICAM-1 – intercelulární adhezní molekula 1; IFN-γ – interferon γ; IL – interleukin; LEMS – Lambertův-Eatonův myastenický syndrom; MAC – membrány atakující komplex; MG – myasthenia gravis; MPP – matrixová metaloproteináza; NO – oxid dusnatý; PM – polymyozitida; TGF-β – transformující růstový faktor β; TNF-α – tumor nekrotizující faktor α; VCAM-1 – vaskulární adhezní molekula 1](https://www.remedia.cz/photo-a-28927---.jpg) Terapeutické působení IVIG je mnohotvárné a komplexní (obr. 1). Zasahuje do většiny složek imunitní regulační sítě. Mezi jeho hlavní účinky patří interference s kostimulačními molekulami, působí jako antiidiotypové protilátky nebo potlačuje tvorbu autoprotilátek. Interferuje s aktivací komplementu a MAC, moduluje expresi a funkci receptorů na makrofázích, potlačuje tvorbu cytokinů, chemokinů a adhezních molekul a snižuje aktivaci, diferenciaci a efektorové funkce T buněk [25–27]. U GBS, MG a LEMS se uplatňuje působení antiidiotypových protilátek nebo suprese protilátkové aktivity [28]. Inhibice vazby komplementu a prevence formace MAC převažuje u GBS, CIDP, MG a DM [29]. Modulace Fc receptorů na makrofázích je patrná u CIDP, GBS a PM a DM [28]. Potlačení tvorby prozánětlivých cytokinů, chemokinů a adhezních molekul na endoteliálních buňkách je zřejmé u zánětlivých demyelinizačních polyneuropatií a myopatií [30].
Terapeutické působení IVIG je mnohotvárné a komplexní (obr. 1). Zasahuje do většiny složek imunitní regulační sítě. Mezi jeho hlavní účinky patří interference s kostimulačními molekulami, působí jako antiidiotypové protilátky nebo potlačuje tvorbu autoprotilátek. Interferuje s aktivací komplementu a MAC, moduluje expresi a funkci receptorů na makrofázích, potlačuje tvorbu cytokinů, chemokinů a adhezních molekul a snižuje aktivaci, diferenciaci a efektorové funkce T buněk [25–27]. U GBS, MG a LEMS se uplatňuje působení antiidiotypových protilátek nebo suprese protilátkové aktivity [28]. Inhibice vazby komplementu a prevence formace MAC převažuje u GBS, CIDP, MG a DM [29]. Modulace Fc receptorů na makrofázích je patrná u CIDP, GBS a PM a DM [28]. Potlačení tvorby prozánětlivých cytokinů, chemokinů a adhezních molekul na endoteliálních buňkách je zřejmé u zánětlivých demyelinizačních polyneuropatií a myopatií [30].
Guillainův-Barréův syndrom
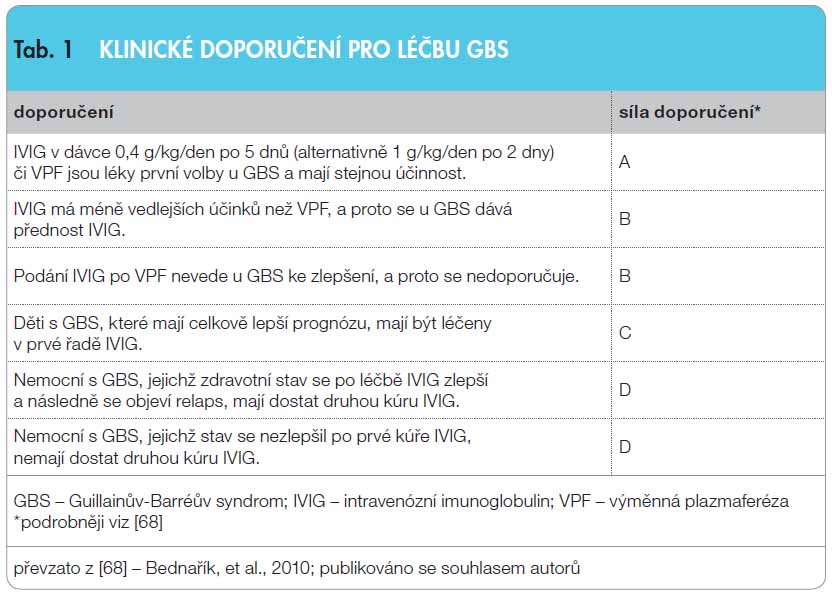 GBS je nejčastějším akutním onemocněním periferních nervů. Klinicky se projevuje rychlou a variabilní manifestací paretických projevů s poruchami citlivosti a někdy i autonomních funkcí. Rozvoj trvá 2, maximálně 4 týdny. Průběh je monofázický, recidivy se vyskytují ve 3–5 %. Přibližně jedna čtvrtina nemocných je ohrožena respirační insuficiencí. Diagnózu je možné potvrdit elektromyografickým vyšetřením a nálezem proteinocytologické disociace v likvoru. Prognóza onemocnění závisí na kvalitě ošetřovatelské péče, úrovni neurointenzivní péče a na adekvátní rehabilitaci [31]. Výměnná plazmaferéza (VPF) urychlí navození klinické remise, ale efekt léčby IVIG je srovnatelný s VPF jak v klinickém zlepšení, tak i v celkové době úzdravy. Dřívější kontrolované klinické studie zjistily, že podávání IVIG v dávce 0,4 g/kg po dobu 5 dnů zlepší motorické funkce a urychlí klinický účinek u signifikantně většího množství pacientů s GBS než VPF [32]. Ačkoliv oba léčebné postupy prokazují jednoznačnou účinnost, jejich kombinace již tento efekt nezvyšuje. U velké kontrolované studie (n = 386), která porovnávala 5denní podávání IVIG v dávce 0,4 g/kg/den, VPF a kombinaci IVIG a VPF, nebyl pozorován signifikantní rozdíl mezi léčenými skupinami při hodnocení disability 4 týdny po randomizaci [33]. Výsledky další studie neprokázaly větší přínos kombinace methylprednisolonu s IVIG proti léčbě IVIG samotným [34]. Další klinické studie potvrdily srovnatelný efekt IVIG a VPF u pacientů s GBS [35, 36]. Podávání kortikoidů (KS) není u GBS účinné. Diskuse o optimální dávce IVIG byly osvětleny francouzskou multicentrickou kontrolovanou studií podávající IVIG v denním režimu 0,4 g/kg po dobu 3 dnů a 0,4 g/kg po dobu 6 dnů. Čas nutný k dosažení samostatné chůze byl signifikantně kratší u druhé skupiny (131 vs. 84 dnů). Stejně tak délka umělé plicní ventilace byla u ventilovaných pacientů významně kratší [37]. IVIG má tedy vyšší účinnost při podávání v minimální dávce 2 g/kg, zejména u pacientů s ventilační podporou. Při monofázickém průběhu nebo při minimálním léčebném efektu IVIG není odůvodněno podávání další kúry, ačkoliv v jedné nekontrolované studii byla popsána účinnost opakovaného podání IVIG [38]. Klinické doporučení pro léčbu GBS je patrné z tab. 1.
GBS je nejčastějším akutním onemocněním periferních nervů. Klinicky se projevuje rychlou a variabilní manifestací paretických projevů s poruchami citlivosti a někdy i autonomních funkcí. Rozvoj trvá 2, maximálně 4 týdny. Průběh je monofázický, recidivy se vyskytují ve 3–5 %. Přibližně jedna čtvrtina nemocných je ohrožena respirační insuficiencí. Diagnózu je možné potvrdit elektromyografickým vyšetřením a nálezem proteinocytologické disociace v likvoru. Prognóza onemocnění závisí na kvalitě ošetřovatelské péče, úrovni neurointenzivní péče a na adekvátní rehabilitaci [31]. Výměnná plazmaferéza (VPF) urychlí navození klinické remise, ale efekt léčby IVIG je srovnatelný s VPF jak v klinickém zlepšení, tak i v celkové době úzdravy. Dřívější kontrolované klinické studie zjistily, že podávání IVIG v dávce 0,4 g/kg po dobu 5 dnů zlepší motorické funkce a urychlí klinický účinek u signifikantně většího množství pacientů s GBS než VPF [32]. Ačkoliv oba léčebné postupy prokazují jednoznačnou účinnost, jejich kombinace již tento efekt nezvyšuje. U velké kontrolované studie (n = 386), která porovnávala 5denní podávání IVIG v dávce 0,4 g/kg/den, VPF a kombinaci IVIG a VPF, nebyl pozorován signifikantní rozdíl mezi léčenými skupinami při hodnocení disability 4 týdny po randomizaci [33]. Výsledky další studie neprokázaly větší přínos kombinace methylprednisolonu s IVIG proti léčbě IVIG samotným [34]. Další klinické studie potvrdily srovnatelný efekt IVIG a VPF u pacientů s GBS [35, 36]. Podávání kortikoidů (KS) není u GBS účinné. Diskuse o optimální dávce IVIG byly osvětleny francouzskou multicentrickou kontrolovanou studií podávající IVIG v denním režimu 0,4 g/kg po dobu 3 dnů a 0,4 g/kg po dobu 6 dnů. Čas nutný k dosažení samostatné chůze byl signifikantně kratší u druhé skupiny (131 vs. 84 dnů). Stejně tak délka umělé plicní ventilace byla u ventilovaných pacientů významně kratší [37]. IVIG má tedy vyšší účinnost při podávání v minimální dávce 2 g/kg, zejména u pacientů s ventilační podporou. Při monofázickém průběhu nebo při minimálním léčebném efektu IVIG není odůvodněno podávání další kúry, ačkoliv v jedné nekontrolované studii byla popsána účinnost opakovaného podání IVIG [38]. Klinické doporučení pro léčbu GBS je patrné z tab. 1.
Chronická zánětlivá demyelinizační polyneuropatie
Klinicky je CIDP charakterizována progresivní symetrickou parézou, poruchou citlivosti a areflexií. Na rozdíl od GBS, který probíhá monofázicky, CIDP plynule progreduje, nebo relabuje. Manifestuje se motorickým a/nebo senzitivním deficitem na více než jedné končetině, progredujícím nebo relabujícím po dobu více než 2 měsíců. K udržení klinické remise je nutné, aby léčba byla dlouhodobá. V diagnostice se používají elektromyografická a laboratorní kritéria [39]. Kontrolované studie prokazují srovnatelný léčebný efekt KS, IVIG i VPF. První jednoduše zaslepená klinická studie z roku 1994 prokázala u pacientů s CIDP srovnatelný efekt IVIG a VPF. Spočívala v týdenním podávání VPF nebo IVIG v dávce 0,2–0,4 g/kg týdně [40]. Podobné výsledky přinesla studie porovnávající IVIG s placebem [41]. Jedna z mnohých randomizovaných kontrolovaných studií srovnávala efekt perorálně podávaného prednisonu (postupně ve sniž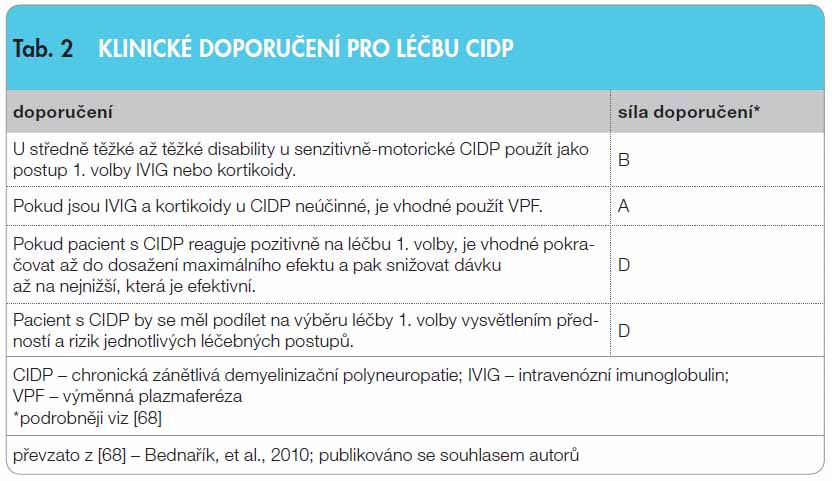 ující se dávce od 60 mg do 10 mg denně) s 2denní aplikací IVIG v dávce 1 g/kg denně. Oba typy léčby přinesly signifikantní zlepšení disability po 2 týdnech, přičemž kvalita života u nemocných, kteří dostávali IVIG, byla lepší a byl u nich zaznamenán menší výskyt nežádoucích účinků [42]. Pozici IVIG v hierarchii léčby CIDP se snažila objasnit 3letá placebem kontrolovaná studie léčebně „naivních“ pacientů. Pacientům byl podán IVIG v dávce 1 g/kg nebo placebo v 1., 2. a 21. den léčby. Rozdíl ve svalové síle byl pozorován již 10. den, s tím, že 42. den byl léčebný efekt signifikantně lepší než při podávání placeba [43]. V roce 2008 byla ukončena studie ICE, která sledovala efekt IVIG podávaného každé 3 týdny po dobu 24 týdnů oproti placebu. Ve studii bylo zařazeno 117 subjektů. Studie prokázala, že dlouhodobější podávání IVIG výrazně redukuje riziko časných relapsů. Klinické výsledky byly lepší, pokud se s léčbou začalo co nejdříve [44, 45]. Výsledky potvrdila studie Latova a kol., přičemž výskyt nežádoucích účinků u pacientů léčených IVIG byl nižší než 0,8 % [46]. Z nedávno publikované metaanalýzy dosud provedených studií vyplývá, že IVIG je vhodný jako léčba první volby zejména v časných fázích těžších forem CIDP. Úvodní dávka je 2 g/kg rozdělená ve 2 nebo 5 dnech. Pokud dojde k signifikantnímu zlepšení, je doporučeno opakovat podání IVIG v intervalu 3 týdnů v dávce 0,4 g/kg [47, 48]. IVIG má nezastupitelné místo v léčbě CIDP asociované s diabetem [49]. Klinické doporučení je uvedeno v tab. 2.
ující se dávce od 60 mg do 10 mg denně) s 2denní aplikací IVIG v dávce 1 g/kg denně. Oba typy léčby přinesly signifikantní zlepšení disability po 2 týdnech, přičemž kvalita života u nemocných, kteří dostávali IVIG, byla lepší a byl u nich zaznamenán menší výskyt nežádoucích účinků [42]. Pozici IVIG v hierarchii léčby CIDP se snažila objasnit 3letá placebem kontrolovaná studie léčebně „naivních“ pacientů. Pacientům byl podán IVIG v dávce 1 g/kg nebo placebo v 1., 2. a 21. den léčby. Rozdíl ve svalové síle byl pozorován již 10. den, s tím, že 42. den byl léčebný efekt signifikantně lepší než při podávání placeba [43]. V roce 2008 byla ukončena studie ICE, která sledovala efekt IVIG podávaného každé 3 týdny po dobu 24 týdnů oproti placebu. Ve studii bylo zařazeno 117 subjektů. Studie prokázala, že dlouhodobější podávání IVIG výrazně redukuje riziko časných relapsů. Klinické výsledky byly lepší, pokud se s léčbou začalo co nejdříve [44, 45]. Výsledky potvrdila studie Latova a kol., přičemž výskyt nežádoucích účinků u pacientů léčených IVIG byl nižší než 0,8 % [46]. Z nedávno publikované metaanalýzy dosud provedených studií vyplývá, že IVIG je vhodný jako léčba první volby zejména v časných fázích těžších forem CIDP. Úvodní dávka je 2 g/kg rozdělená ve 2 nebo 5 dnech. Pokud dojde k signifikantnímu zlepšení, je doporučeno opakovat podání IVIG v intervalu 3 týdnů v dávce 0,4 g/kg [47, 48]. IVIG má nezastupitelné místo v léčbě CIDP asociované s diabetem [49]. Klinické doporučení je uvedeno v tab. 2.
Multifokální motorická neuropatie
Toto onemocnění se klinicky manifestuje pomalu progredující asymetrickou, převážně distální slabostí s atrofiemi, bez poruchy citlivosti. Diagnózu je možné ověřit elektromyografickým vyšetřením s nálezem vícečetných parciálních bloků vedení motorických nervů, užitečné je vyšetření protilátek proti gangliosidům GM1 [50]. Zatímco CIDP reaguje na léčbu KS, u MMN tomu tak není, KS dokonce mohou onemocnění zhoršit. Více randomizovaných, kontrolovaných, dvojitě zaslepených studií prokázalo, že IVIG zlepšuje svalovou sílu, skóre disability a elektromyografické parametry [5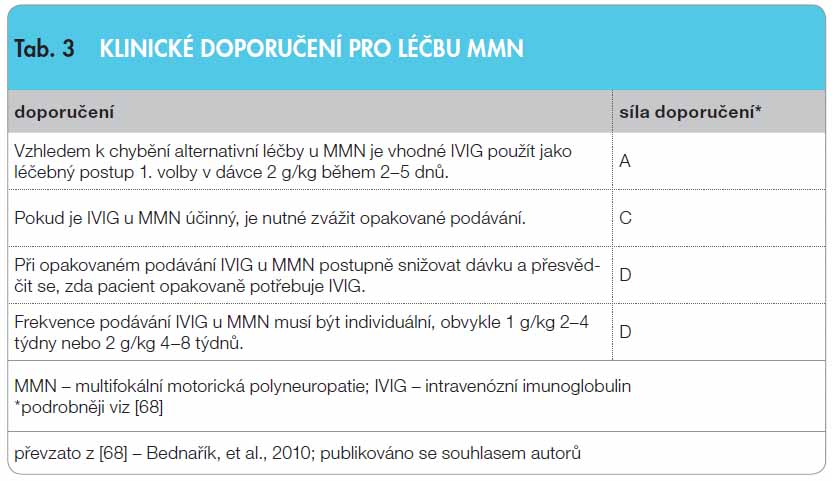 1–54]. Malá kontrolovaná studie prokázala u 11 nemocných s MMN efekt při dlouhodobé léčbě. Po iniciální dávce 0,4 g/kg/den po dobu 5 dnů byla aplikována dávka IVIG 0,4 g/kg jednou týdně po dobu jednoho roku, eventuálně dle klinického nálezu i déle. Svalová síla se zlepšila průměrně po 3 týdnech léčby, ale mírně klesala během sledovaného období. Je zajímavé, že se elektrofyziologické změny patrné při zlepšení (remyelinizace nebo reinervace) či při zhoršení (demyelinizace nebo axonální poškození) objevovaly paralelně v různých nervech, zatímco kondukční blok mizel v jedněch, ale objevoval se v jiných. Tyto nálezy nasvědčují tomu, že je v některých případech nutná dlouhodobá udržovací léčba. V 6měsíční observační studii se dokonce dávka IVIG postupně navyšovala z 0,5 g/kg/měsíc až na 2 g/kg/měsíc dle klinických a elektromyografických nálezů [55]. Klinické doporučení uvádí tab. 3.
1–54]. Malá kontrolovaná studie prokázala u 11 nemocných s MMN efekt při dlouhodobé léčbě. Po iniciální dávce 0,4 g/kg/den po dobu 5 dnů byla aplikována dávka IVIG 0,4 g/kg jednou týdně po dobu jednoho roku, eventuálně dle klinického nálezu i déle. Svalová síla se zlepšila průměrně po 3 týdnech léčby, ale mírně klesala během sledovaného období. Je zajímavé, že se elektrofyziologické změny patrné při zlepšení (remyelinizace nebo reinervace) či při zhoršení (demyelinizace nebo axonální poškození) objevovaly paralelně v různých nervech, zatímco kondukční blok mizel v jedněch, ale objevoval se v jiných. Tyto nálezy nasvědčují tomu, že je v některých případech nutná dlouhodobá udržovací léčba. V 6měsíční observační studii se dokonce dávka IVIG postupně navyšovala z 0,5 g/kg/měsíc až na 2 g/kg/měsíc dle klinických a elektromyografických nálezů [55]. Klinické doporučení uvádí tab. 3.
Myasthenia gravis
Pro MG je typická kolísající slabost a únavnost predilekčně postižených svalů (extraokulární, orofaryngeální, šíjové, pletencové a respirační). Onemocnění probíhá v exacerbacích a remisích, některé z nich mohou vyústit do 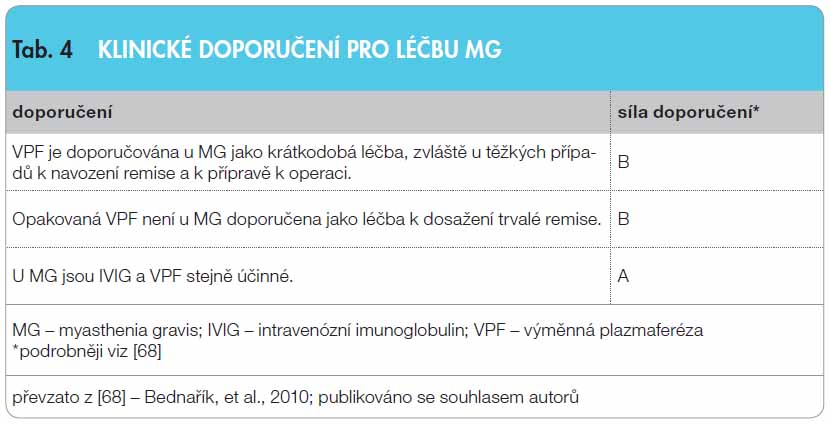 myastenické krize s respiračním selháním [56]. Dlouhodobou léčbou první volby je podávání inhibitorů cholinesterázy, ve většině případů je nutné je kombinovat s KS, eventuálně spolu s imunosupresivy. U mladších nemocných je indikována thymektomie. U exacerbací MG je prováděna VPF nebo je podáván IVIG. První randomizovaná studie prokázala již v roce 1997 srovnatelný efekt VPF a IVIG. V této studii byl podáván IVIG 87 pacientům s exacerbací MG v dávce 0,4 g/kg/den po dobu 3 nebo 5 dnů. Klinický efekt a pokles hladiny protilátek proti acetylcholinovému receptoru byl podobný [57]. V dalších studiích byl dokumentován efekt proti placebu. Jiná studie neprokázala signifikantní rozdíl v celkové dávce 1 g/kg a 2 g/kg. Údaje o efektu dlouhodobé léčby podáváním IVIG u pacientů s MG rezistentních k imunosupresivní léčbě jsou doloženy kazuistikami, nikoliv však kontrolovanou studií [58, 59]. V tab. 4 je shrnuto doporučení pro léčbu.
myastenické krize s respiračním selháním [56]. Dlouhodobou léčbou první volby je podávání inhibitorů cholinesterázy, ve většině případů je nutné je kombinovat s KS, eventuálně spolu s imunosupresivy. U mladších nemocných je indikována thymektomie. U exacerbací MG je prováděna VPF nebo je podáván IVIG. První randomizovaná studie prokázala již v roce 1997 srovnatelný efekt VPF a IVIG. V této studii byl podáván IVIG 87 pacientům s exacerbací MG v dávce 0,4 g/kg/den po dobu 3 nebo 5 dnů. Klinický efekt a pokles hladiny protilátek proti acetylcholinovému receptoru byl podobný [57]. V dalších studiích byl dokumentován efekt proti placebu. Jiná studie neprokázala signifikantní rozdíl v celkové dávce 1 g/kg a 2 g/kg. Údaje o efektu dlouhodobé léčby podáváním IVIG u pacientů s MG rezistentních k imunosupresivní léčbě jsou doloženy kazuistikami, nikoliv však kontrolovanou studií [58, 59]. V tab. 4 je shrnuto doporučení pro léčbu.
Lambertův-Eatonův myastenický syndrom
Pacienti s Lambertovým-Eatonovým myastenickým syndromem trpí proximální svalovou slabostí, někdy i očními a bulbárními příznaky a vegetativní dysfunkcí s charakteristickým elektromyografickým nálezem. V 60 % jde o paraneoplastický syndrom asociovaný s malobuněčným karcinomem plic [60]. Většina nemocných odpovídá na léčbu KS a imunosupresivy, někteří na symptomatickou léčbu 3,4-amidopyridinem. Účinnost VPF i IVIG byla prokázána u LEMS jen jednou randomizovanou studií kontrolovanou placebem [61]. V případě inoperabilního tumoru je indikace IVIG diskutabilní.
Zánětlivé myopatie
PM a DM jsou charakteristické subakutně se rozvíjející, většinou symetrickou proximální svalovou slabostí. Některé formy mohou probíhat akutně. PM postihuje převážně dospělé jedince. Ve svalové biopsii se nacházejí endomysiální zánětlivé infiltráty. U DM se svalové postižení kombinuje s výskytem kožních změn (heliotropní exantém, Gottronovy změny), ve svalové biopsii se nalézají perivaskulární a perimysiální infiltráty. Zánětlivé myopatie se mohou vyskytovat ve formě paraneoplastického onemocnění, nebo jako over lap (překryvný) syndrom, v asociaci s jinými autoi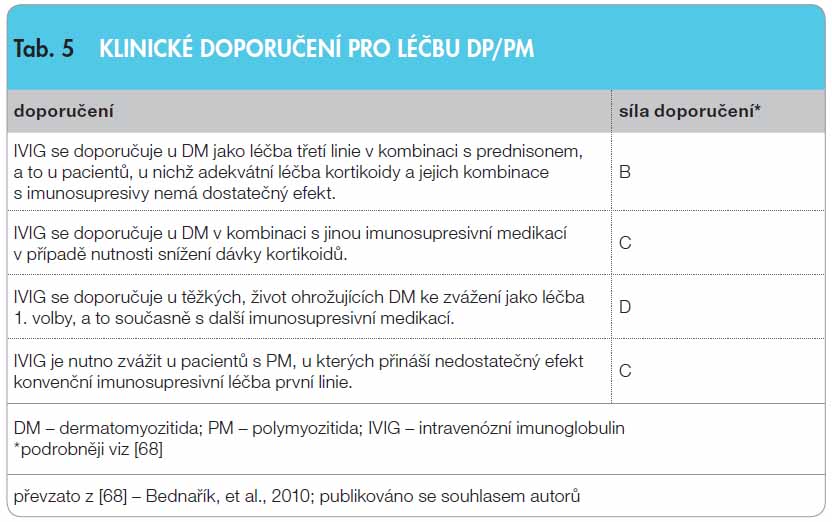 munitními chorobami. Standardní léčba spočívá v podání KS, často v kombinaci s imunosupresivy [62]. IVIG je považován za lék 2.–3. volby. V literatuře jsou k dispozici 2 randomizované kontrolované studie, 9 studií otevřených a 3 retrospektivní, jež analyzují velmi příznivý terapeutický efekt u 308 dospělých pacientů s DM a PM. Běžná dávka je 2 g/kg ve 2–5 kúrách. Efekt léčby oproti placebu je patrný v klinických parametrech, v poklesu hladiny kreatinkinázy a ve snížení dávek KS. IVIG představuje léčbu volby u akutních DM/PM nebo u nemocných, u kterých jsou KS kontraindikovány [63]. U myozitidy s inkluzními tělísky nebyl léčebný efekt IVIG prokázán. Klinické doporučení je uvedeno v tab. 5.
munitními chorobami. Standardní léčba spočívá v podání KS, často v kombinaci s imunosupresivy [62]. IVIG je považován za lék 2.–3. volby. V literatuře jsou k dispozici 2 randomizované kontrolované studie, 9 studií otevřených a 3 retrospektivní, jež analyzují velmi příznivý terapeutický efekt u 308 dospělých pacientů s DM a PM. Běžná dávka je 2 g/kg ve 2–5 kúrách. Efekt léčby oproti placebu je patrný v klinických parametrech, v poklesu hladiny kreatinkinázy a ve snížení dávek KS. IVIG představuje léčbu volby u akutních DM/PM nebo u nemocných, u kterých jsou KS kontraindikovány [63]. U myozitidy s inkluzními tělísky nebyl léčebný efekt IVIG prokázán. Klinické doporučení je uvedeno v tab. 5.
Nežádoucí účinky
Bezpečnost léčby se zvyšuje moderními technologiemi výroby IVIG. Například přenos infekčních agens se prakticky nevyskytuje. Nežádoucí účinky jsou poměrně vzácné. U necelého 1 % pacientů se může během infuze objevit třesavka, zvýšená teplota, bolesti hlavy, nauzea a vomitus. Anafylaktický šok je raritní. Další příznaky jsou důsledkem zvýšené hladiny IgG (reverzibilní renální poškození, akutní hemolýza, neutropenie, trombózy, aseptická meningitida) [64].
Závěr
Léčba nervosvalových autoimunitních onemocnění je relativně bezpečná a efektivní. Výrazným způsobem zlepšila prognózu některých závažných onemocnění, např. MMN a MG. Vhledem k tomu, že jde o poměrně nákladnou terapii, je třeba ji indikovat na základě medicíny založené na důkazech. Klinická doporučení se opírají o metaanalýzu klinických studií, výsledky Cochranovy databáze, doporučení Evropské asociace neurologických společností (EFNS), Americké akademie urgentní medicíny (AANEM), Americké neurologické akademie (AAN) a Českého standardu pro léčbu pacientů s autoimunitními nervosvalovými onemocněními intravenózním lidským imunoglobulinem a výměnnou plazmaferézou [65–68].
Seznam použité literatury
- [1] Montomoli C, Citterio A, Piccolo G, et al. Epidemiology and geographical variation of myasthenia gravis in the province of Pavia, Italy. Neuroepidemiology 2012; 38: 100–105.
- [2] Laughin RS, Dyck PJ, Melton LJ, et al. Incidence and prevalence of CIDP and the association of diabetes mellitus. Neurology 2009; 73: 39–45.
- [3] Bednařík J. Multifokální motorická neuropatie. Neurol prax 2006; 7: 32–34.
- [4] Sejvar JJ, Baughman AL, Wise M, et al. Population incidence of Guillain-Barré syndrome: A systematic review and meta-analysis. Neuroepidemiology 2011; 36: 123–133.
- [5] Špalek P. Intravenózny imunoglobulín v liečbe autoimunitných neurologických ochorení. Neurol prax 2011; 12: 398–402.
- [6] Gold R, Dalakas MC, Toyka KV. Immunotherapy in autoimmune neuromuscular disorders. Lancet Neurol 2003; 2: 22–32.
- [7] Dalakas MC. Basic aspects of neuroimmunology as they relate to immunotherapeutic targets: present and future prospects. Ann Neurol 1995; 37 (suppl 1): S2–S13.
- [8] Karlsen AE, Dyrberg T. Molecular mimicry between non-self, modified self and self in autoimmunity. Semin Immunol 1998; 10: 25–34.
- [9] Hughes RA, Hadden RD, Gregson NA, et al. Pathogenesis of Guillain-Barre´ syndrome. J Neuroimmunol 1999; 100: 74–97.
- [10] Yuki N. Infectious origins of, and molecular mimicry in Guillain-Barre´ and Fisher syndromes. Lancet Infect Dis 2001; 1: 29–37.
- [11] Ramchandren S, Lisak RP. The immunopathogenesis of Guillain-Barré syndrome. Clin Adv Hematol Oncol 2010; 8: 203–206.
- [12] Shahrizaila N, Yuki N. Antiganglioside antibodies in Guillain-Barré syndrome and its related conditions. Expert Rev Neurother 2011; 11: 1305–1313.
- [13] Mori M, Kuwabara S, Yuki N. Fisher syndrome: clinical features, immunopathogenesis and management. Expert Rev Neurother 2012; 12: 39–51.
- [14] Kieseier BC, Dalakas MC, Hartung HP. Immune mechanisms in chronic inflammatory demyelinating neuropathy. Neurology 2002; 59 (suppl 6): S7–S12.
- [15] Rezania K, Gundogdu B, Soliven B. Pathogenesis of chronic inflammatory demyelinating polyradiculoneuropathy. Front Biosci 2004; 9: 939–945.
- [16] Nobile-Orazio E. Multifocal motor neuropathy. J Neuroimmunol 2001; 115: 4–18.
- [17] Oh SJ, LaGanke C, Powers R, et al. Multifocal motor sensory demyelinating neuropathy: inflammatory demyelinating polyradiculoneuropathy. Neurology 2005; 65: 1639–1642.
- [18] Hafer-Macko C, Hsieh ST, Li CY, et al. Acute motor axonal neuropathy: an antibody-mediated attack on axolemma. Ann Neurol 1996; 40: 635–644.
- [19] Hafer-Macko CE, Sheikh KA, Li CY, et al. Immune attack on the Schwann cell surface in acute inflammatory demyelinating polyneuropathy. Ann Neurol 1996; 39: 625–635.
- [20] Dalakas MC. Molecular immunology and genetics of inflammatory muscle diseases. Arch Neurol 1998; 55: 1509–1512.
- [21] Dalakas MC. The molecular and cellular pathology of inflammatory muscle diseases. Curr Opin Pharmacol 2001; 1: 300–306.
- [22] Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet 2003; 362: 971–982.
- [23] Meriggioli MN, Sanders DB. Autoimmune myasthenia gravis: emerging clinical and biological heterogeneity. Lancet Neurol 2009; 8: 475–490.
- [24] Takamori M, Maruta T, Komai K. Lambert-Eaton myasthenic syndrome as an autoimmune calciumchannelopathy. Neurosci Res 2000; 36: 183–191.
- [25] Dalakas MC. Intravenous immunoglobulin in the treatment of autoimmune neuromuscular diseases: present status and practical therapeutic guidelines. Muscle Nerve 1999; 22: 1479–1497.
- [26] Bayry J, Lacroix-Desmazes S, Carbonneil C, et al. Inhibition of maturation and function of dendritic cells by intravenous immunoglobulin. Blood 2003; 101: 758–765.
- [27] Quick A, Tandan R. Mechanisms of action of intravenous immunoglobulin in inflammatory muscle disease. Curr Rheumatol Rep 2011; 13: 192–198.
- [28] Shahrizaila N, Yuki N. The role of immunotherapy in Guillain-Barré syndrome: understanding the mechanism of action. Expert Opin Pharmacother 2011; 12: 1551–1560.
- [29] Basta M, Illa I, Dalakas MC. Increased in vitro uptake of the complement C3b in the serum of patients with Guillain-Barre´ syndrome, myasthenia gravis and dermatomyositis. J Neuroimmunol 1996; 71: 227–229.
- [30] Lehmann HC, Hartung HP. Plasma exchange and intravenous immunoglobulins: mechanism of action in immune-mediated neuropathies. J Neuroimmunol 2011; 231: 61–69.
- [31] Winer JB. Guillain-Barré syndrome: clinical variants and their pathogenesis. J Neuroimmunol 2011; 231: 70–72.
- [32] Van der Meche FG, Schmitz PI. A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain-Barre´ syndrome. N Engl J Med 1992; 326: 1123–1129.
- [33] Plasma Exchange/Sandoglobulin Guillain- -Barré Syndrome Trial Group. Plasma Exchange/ Sandoglobulin Guillain-Barre´ Syndrome Trial Group. Randomised trial of plasma exchange, intravenous immunoglobulin, and combined treatments in Guillain-Barre´ syndrome. Lancet 1997; 349: 225–230.
- [34] van Koningsveld R, Schmitz PI, van der Meche´ FG, et al. Effect of methylprednisolone when addend to standard treatment with intravenous imunoglobulin for Guillain-Barre´ syndrome: randomized trial. Lancet 2004; 363: 192–196.
- [35] Hughes RA, Swan AV, Raphaël JC, et al. Immunotherapy for Guillain-Barré syndrome: a systematic review. Brain 2007; 130 (Pt 9): 2245–2257.
- [36] Hughes RA, Swan AV, van Doorn PA. Intravenous immunoglobulin for Guillain-Barré syndrome. Cochrane Database Syst Rev 2010; 16;: CD002063.
- [37] Raphael JC, Chevret S, Harboun M, Jars- -Guincestre MC. Intravenous immune globulins in patients with Guillain-Barre´ syndrome and contraindications to plasma exchange: 3 days versus 6 days. J Neurol Neurosurg Psychiatry 2001; 71: 235–238.
- [38] Farcas P, Avnun L, Frisher S, et al. Efficacy of repeated intravenous immunoglobulin in severe unresponsive Guillain-Barre´ syndrome. Lancet 1997; 350: 1747.
- [39] Dalakas MC. Advances in the diagnosis, pathogenesis and treatment of CIDP. Nat Rev Neurol 2011; 16; 7: 507–517.
- [40] Dyck PJ, Litchy WJ, Kratz KM, et al. A plasma exchange versus immune globulin infusion trial in chronic inflammatory demyelinating polyradiculoneuropathy. Ann Neurol 1994; 36: 838–845.
- [41] Hahn AF, Bolton CF, Zochodne D, Feasby TE. Intravenous immunoglobulin treatment in chronic inflammatory demyelinating polyneuropathy: a double blind, placebo-controlled, cross-over study. Brain 1996; 119: 1067–1077.
- [42] Hughes R, Bensa S, Willison H, et al. Randomized controlled trial of intravenous immunoglobulin versus oral prednisolone in chronic inflammatory demyelinating polyradiculoneuropathy. Ann Neurol 2001; 50: 195–201.
- [43] Barohn RJ, Freimer ML, et al. Randomized controlled trial of IVIg in untreated chronic inflammatory demyelinating polyradiculoneuropathy. Neurology 2001; 56: 445–449.
- [44] Hughes RA, Donofrio P, Bril, et al. ICE Study Group.Intravenous immune globulin (10% caprylate- chromatography purified) for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy (ICE study): a randomised placebo-controlled trial. Lancet Neurol 2008; 7: 136–144.
- [45] Hughes RA. Intravenous immunoglobulin for chronic inflammatory demyelinating polyradiculoneuropathy: the ICE trial. Expert Rev Neurother 2009; 9: 789–795.
- [46] Latov N, Deng C, Dalakas MC, et al. IGIV-C CIDP Efficacy (ICE) Study Group. Timing and course of clinical response to intravenous immunoglobulin in chronic inflammatory demyelinating polyradiculoneuropathy. Neurology 2010; 67: 802–807.
- [47] Gaebel K, Blackhouse G, Campbell K, et al. Intravenous immunoglobulin for the treatment of chronic inflammatory demyelinating polyradiculoneuropathy: a systematic review and meta- analysis. Open Med 2010; 4: e154–e166.
- [48] Mahdi-Rogers M, Swan AV, van Doorn PA, Hughes RA. Immunomodulatory treatment other than corticosteroids, immunoglobulin and plasma exchange for chronic inflammatory demyelinating polyradiculoneuropathy. Cochrane Database Syst Rev 2010 10; 11: CD003280.
- [49] Jann S, Bramerio MA, Facchet ti D, Sterzi R. Intravenous immunoglobulin is effective in patients with diabetes and with chronic inflammatory demyelinating polyneuropathy: long term follow-up. J Neurol Neurosurg Psychiatry 2009; 80: 70–73.
- [50] Meuth SG, Kleinschnitz C. Multifocal motor neuropathy: update on clinical characteristics, pathophysiological concepts and therapeutic options. Eur Neurol 2010; 63: 193–204.
- [51] Van den Berg LH, Kerkhoff H, Oey PL, et al. Treatment of multifocal motor neuropathy with high dose intravenous immunoglobulins: a double blind, placebo controlled study. J Neurol Neurosurg Psychiatry 1995; 59: 248–252.
- [52] Azulay JP, Blin O, Pouget J, et al. Intravenous immunoglobulin treatment in patients with motor neuron syndromes associated with anti-GM1 antibodies: a double-blind, placebo-controlled study. Neurology 1994; 44: 429–432.
- [53] Federico P, Zochodne DW, Hahn AF, et al. Multifocal motor neuropathy improved by IVIg: randomized, double-blind, placebo-controlled study. Neurology 2000; 55 :1256–1262.
- [54] van Schaik IN, Léger JM. Nobile-Orazio E, et al. European Federation of Neurological Societies/ Peripheral Nerve Society guideline on management of multifocal motor neuropathy. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society – first revision. J Peripher Nerv Syst 2010; 15: 295–301.
- [55] Baumann A, Hess CW, Sturzenegger M. IVIg dose increase in multifocal motor neuropathy: a prospective six month follow-up. J Neurol 2009; 256: 608–614.
- [56] Piťha J. a kol. Myasthenia gravis a ostatní poruchy nervosvalového přenosu. Praha, Maxdorf 2010, 367 s.
- [57] Gajdos P, Chevret S, Clair B, et al. Clinical trial of plasma exchange and highdose intravenous immunoglobulin in myasthenia gravis. Ann Neurol 1997; 41: 789–796.
- [58] Barth D, Nabavi Nouri M, Ng E, et al. Comparison of IVIg and PLEX in patients with myasthenia gravis. Neurology 2011; 76: 2017–2023.
- [59] Zinman L, Bril V. IVIG treatment for myasthenia gravis: effectiveness, limitations, and novel therapeutic strategies. Ann N Y Acad Sci 2008; 1132: 264–270.
- [60] Titulaer MJ, Lang B, Verschuuren JJ. Lambert- -Eaton myasthenic syndrome: from clinical characteristics to therapeutic strategies. Lancet Neurol 2011; 10: 1098–1107.
- [61] Keogh M, Sedehizadeh S, Maddison P. Treatment for Lambert-Eaton myasthenic syndrome. Cochrane Database Syst Rev 2011; 16: CD003279.
- [62] Pestronk A. Acquired immune and inflammatory myopathies: pathologic classification. Curr Opin Rheumatol 2011; 23: 595–604.
- [63] Wang DX, Shu XM, Tian XL, et al. Intravenous immunoglobulin therapy in adult patients with polymyositis/ dermatomyositis: a systematic literature review. Clin Rheumatol 2012; 31: 801–806.
- [64] Rezaei N, Abolhassani H, Aghamohammadi A, Ochs HD. Indications and safety of intravenous and subcutaneous immunoglobulin therapy. Expert Rev Clin Immunol 2011; 7: 301–316.
- [65] Elovaara I, Apostolski S, van Doorn P, et al. EFNS guidelines for the use of intravenous immunoglobulin in treatment of neurological diseases: EFNS task force on the use of intravenous immunoglobulin in treatment of neurological diseases. Eur J Neurol 2008; 15: 893–908.
- [66] Donofrio PD, Berger A, Brannagan TH 3rd, et al. Consensus statement: the use of intravenous immunoglobulin in the treatment of neuromuscular conditions report of the AANEM ad hoc committee. Muscle Nerve 2009; 40: 890–900.
- [67] Evidence-based guideline: Intravenous immunoglobulin in the treatment of neuromuscular disorders: Report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Patwa HS, Chaudhry V, Katzberg H, et al. Neurology 2012; 78: 1009–1015.
- [68] Bednařík J, Voháňka S, Ehler E, et al. Standard pro léčbu pacientů s autoimunitními nervosvalovými onemocněními intravenózním lidským imunoglobulinem a výměnnou plazmaferézou. 2010. Dostupné z URL http://www.sopr.cz/ standardy/#definitivni