Léčba Fabryho choroby
Fabryho choroba je dědičné metabolické onemocnění na podkladě mutace genu pro enzym α-galaktosidázu A, který je lokalizován na chromozomu X. Projevuje se hromaděním globotriaosylceramidu s následnou hypertrofickou kardiomyopatií, selháním ledvin, postižením periferního i centrálního nervového systému, kůže, očí i dalších orgánů. V posledním desetiletí došlo k velkému rozvoji poznatků a k zavedení specifické terapie této choroby. Substituce chybějícího enzymu se podává nitrožilně jedenkrát za 2 týdny. Klinické studie prokázaly dobrý efekt, bylo-li s léčbou započato před rozvinutím ireverzibilního postižení. V budoucnu se dočkáme zavedení terapie malými molekulami (chaperony), které prodlužují poločas enzymu, a genetické terapie.
Úvod
![Obr. 1 Gen pro α-galaktosidázu je lokalizován na dlouhém raménku chromozomu X v pozici 22 (páry bází 100, 539, 452 až 100, 549, 606); podle [1] – www.wiki.medpedia.com.](https://www.remedia.cz/photo-a-28917---.jpg) Fabryho choroba je vzácné dědičné onemocnění patřící mezi lysozomální choroby ze střádání. Je způsobeno mutací genu pro enzym α-galaktosidázu A, který se účastní odbourávání glykosfingolipidů. Gen je lokalizován na chromozomu X, jedná se tedy o gonozomálně recesivní dědičnost (obr. 1). Mutace způsobuje absolutní nebo relativní nedostatek enzymu a důsledkem je hromadění globotriaosylceramidu (Gb3) v lysozomech buněk různých tkání a jejich poškození. Existuje několik set těchto mutací, které jsou většinou typické pro jednotlivé rodiny (tzv. privátní mutace). Vzhledem k přítomnosti mutovaného genu na chromozomu X budou všichni mužští potomci postiženého muže zdrávi a všechny ženy zdědí mutaci. Postižené ženy přenášejí mutaci na polovinu potomků obou pohlaví.
Fabryho choroba je vzácné dědičné onemocnění patřící mezi lysozomální choroby ze střádání. Je způsobeno mutací genu pro enzym α-galaktosidázu A, který se účastní odbourávání glykosfingolipidů. Gen je lokalizován na chromozomu X, jedná se tedy o gonozomálně recesivní dědičnost (obr. 1). Mutace způsobuje absolutní nebo relativní nedostatek enzymu a důsledkem je hromadění globotriaosylceramidu (Gb3) v lysozomech buněk různých tkání a jejich poškození. Existuje několik set těchto mutací, které jsou většinou typické pro jednotlivé rodiny (tzv. privátní mutace). Vzhledem k přítomnosti mutovaného genu na chromozomu X budou všichni mužští potomci postiženého muže zdrávi a všechny ženy zdědí mutaci. Postižené ženy přenášejí mutaci na polovinu potomků obou pohlaví.
![Graf 1 Průběh Fabryho choroby v závislosti na čase; podle [17] – Wanner, 2007.](https://www.remedia.cz/photo-a-28918---.jpg) Prevalence choroby se udává 1 případ na 100 tisíc obyvatel. Pravděpodobně je však onemocnění poddiagnostikováno a prevalence bude vyšší (odhaduje se 1 případ na 40 tisíc obyvatel). S chorobou se
Prevalence choroby se udává 1 případ na 100 tisíc obyvatel. Pravděpodobně je však onemocnění poddiagnostikováno a prevalence bude vyšší (odhaduje se 1 případ na 40 tisíc obyvatel). S chorobou se
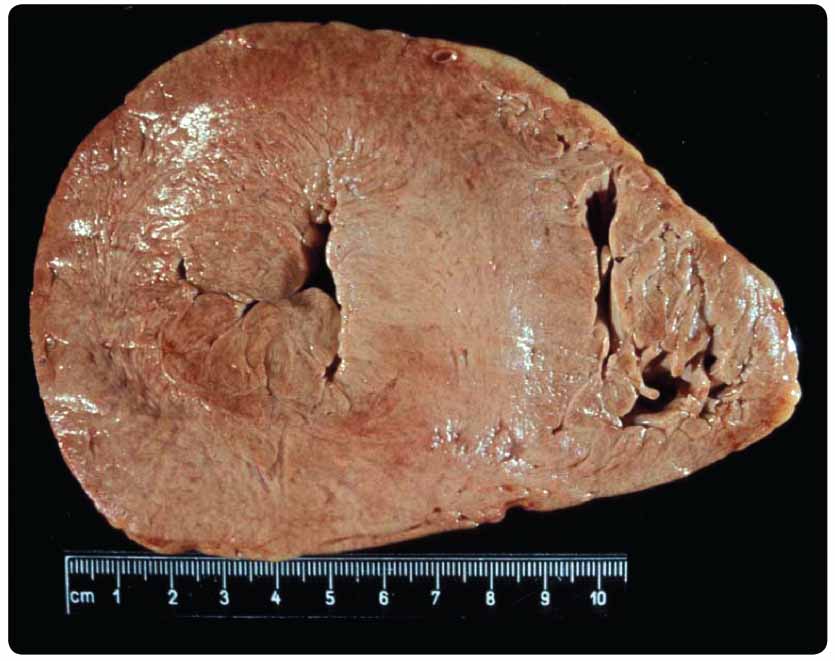
setkáváme ve všech zemích a postihuje všechny lidské rasy. Ke střádání dochází dokonce již intrauterinně. Akumulace Gb3 vede postupně k poškození tkání a následně k orgánovému selhání (graf 1). Klasická forma choroby se projevuje již v dětství postižením periferní nervové soustavy (bolestmi, pálením dlaní a chodidel, gastrointestinálními příznaky), ve druhém a třetím decenniu se přidává postižení srdce (hypertrofická kardiomyopatie, poruchy srdečního rytmu, vzácně i postižení srdečních chlopní) a postižení ledvin, které vede k terminálnímu selhání ledvin (obr.2 a 3).
Častěji se vyskytují také cévní mozkové příhody. Typické je postižení očí, kde nacházíme korneální opacity zvané cornea verticillata a tortuozitu spojivkových i retinálních cév. Postižení zraku zpravidla nevede k vážnějšímu omezení zrakové ostrosti. Postižení kůže se projevuje angiokeratomy, které jsou sice charakteristické, ale nejsou pro Fabryho chorobu specifické. Angiokeratomy jsou typicky lokalizovány na bocích, hýždích, flexorové straně stehen a v genitální krajině. Velmi závažné bývá postižení sluchu, které často vede k úplné hluchotě. Nepříjemné a na terapii rezistentní je vertigo při postižení vestibulárního aparátu. Ženy mohou být rovněž postiženy, přestože je gen lokalizován na chromozomu X. Postižení je u žen většinou mírnější a manifestuje se později. Mezi našimi pacientkami ale není výjimečné ani terminální selhání ledvin ve 3–4. decenniu. U obou pohlaví se mohou vyskytovat také formy, které postihují pouze jeden orgán (např. kardiální nebo renální forma choroby). U takto postižených pacientů bývá přítomna reziduální aktivita α-galaktosidázy A. 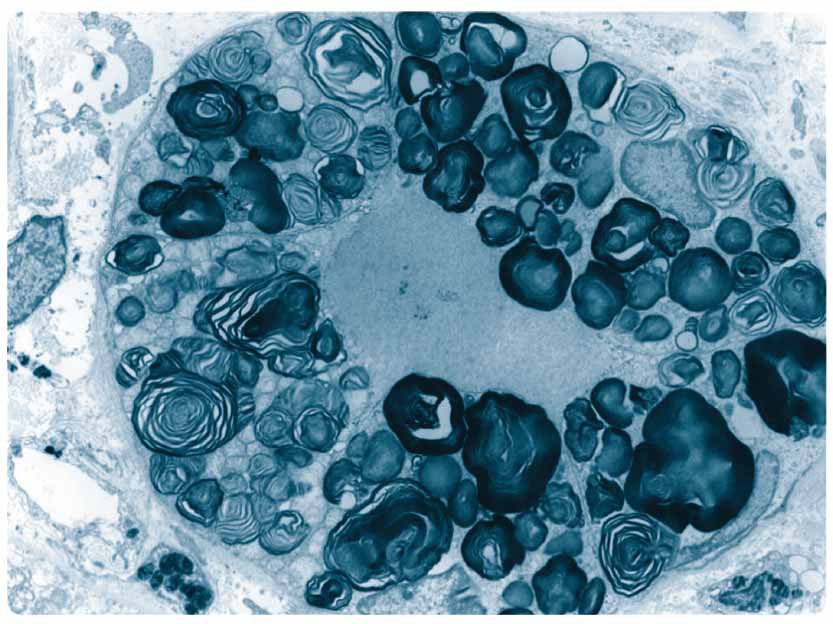 Postižení žen a často také mužů může být velmi variabilní, a to dokonce i v rámci jedné rodiny. Studie zabývající se korelacemi genotypu a fenotypu tuto variabilitu nevysvětlují. U žen může být příčinou lyonizace (náhodná inaktivace jednoho z chromozomů X). Bez léčby má postižení u mužů zpravidla progresivní charakter a vede k závažnému orgánovému postižení. Výzkum se v oblasti léčby zabývá jednak možností snížení produkce substrátu (substrát deprivační-terapie) a jednak možností zvýšení produkce a dostupnosti enzymu. Toho lze docílit transplantací kostní dřeně, pomocí malých molekul (tzv. chaperonů), genovou léčbou a enzym-substituční terapií, která je v současnosti ze všech těchto metod nejúspěšnější.
Postižení žen a často také mužů může být velmi variabilní, a to dokonce i v rámci jedné rodiny. Studie zabývající se korelacemi genotypu a fenotypu tuto variabilitu nevysvětlují. U žen může být příčinou lyonizace (náhodná inaktivace jednoho z chromozomů X). Bez léčby má postižení u mužů zpravidla progresivní charakter a vede k závažnému orgánovému postižení. Výzkum se v oblasti léčby zabývá jednak možností snížení produkce substrátu (substrát deprivační-terapie) a jednak možností zvýšení produkce a dostupnosti enzymu. Toho lze docílit transplantací kostní dřeně, pomocí malých molekul (tzv. chaperonů), genovou léčbou a enzym-substituční terapií, která je v současnosti ze všech těchto metod nejúspěšnější.
Enzym-substituční terapie
Enzym-substituční terapie (ERT, Enzyme Replacement Therapy) je dostupná teprve v posledním desetiletí. Fabryho choroba je po Gaucherově chorobě druhým lysozomálním onemocněním ze střádání, u kterého se podařilo vyrobit a úspěšně používat enzym-substituční terapii. Zprvu se lék podával v rámci klinických studií a po jejich příznivých výsledcích byl schválen ke komerčnímu použití. Na trhu existují dva preparáty, agalsidáza beta a agalsidáza alfa. Léčba se podává v krátké infuzi 1krát za 14 dní. Tolerance léku je zpravidla dobrá, ale mohou se vyskytnout nežádoucí účinky a alergické reakce.
Farmakokinetika
Plazmatická koncentrace léku vrcholí několik minut po ukončení infuze. Poté plazmatická aktivita klesá postupně k nule.Aktivita podaného léku není detekovatelná již 24 hodin po jeho podání. Poločas závisí na dávce a pohybuje se v rozmezí od 45 do 100 minut [2].
Agalsidáza beta i alfa jsou proteiny a jsou štěpeny peptidovou hydrolýzou. Významnější hepatopatie metabolismus léku neovlivňuje a vylučování ledvinami je nevýznamné. Možná tvorba protilátek v experimentu neovlivňovala vychytávání léku buňkami a nemá pravděpodobně vliv na účinnost léčby. Vzhledem k proteinové povaze léku nehrozí ani lékové interakce.
Agalsidáza beta se vyrábí na tkáňových kulturách ovariálních buněk čínských křečků. Doporučená dávka je 1 mg/kg
podávaná 1krát za 14 dní. Agalsidáza alfa se vyrábí na lidských tkáňových kulturách a doporučená dávka je 0,2 mg/kg. Rozdíly ve složení jsou minimální, liší se pouze v glykosylaci (v obsahu kyseliny sialové a manoso-6-fosfátu). V ČR i Evropské unii se používají oba preparáty a jejich cena je obdobná. V USA je schválena zatím pouze agalsidáza beta, u agalsidázy alfa se čeká na dokončení několika probíhajících klinických studií. Léčba je velmi nákladná. V ČR je péče o pacienty s Fabryho chorobou soustředěna do Centra pro Fabryho chorobu na II. interní klinice kardiologie a angiologie VFN a 1. LF UK. Léčbu je možné zahájit jen se souhlasem příslušné pojišťovny a vedení nemocnice po posouzení indikace k léčbě dle vnitřních doporučení.
Účinnost enzym-substituční léčby
ERT prokázala účinnost v tkáňových kulturách i v klinických studiích. Oba léky zvyšovaly aktivitu α-galaktosidázy A v tkáňových kulturách. Klinické studie rovněž prokázaly, že lék dokáže vyčistit tkáně od Gb3 a zpomalit progresi onemocnění. V četných klinických studiích se potvrdilo, že léčba dokáže zpomalit nebo zastavit zhoršování renálních funkcí, zpomalit progresi či navodit regresi hypertrofie myokardu, zabránit kardiovaskulárním komplikacím a zlepšit kvalitu života. Avšak k posouzení mortality jsou naše zkušenosti zatím krátké. Problémem posuzování účinnosti je také malý počet pacientů a dále značně variabilní klinický obraz. Problém malého počtu pacientů v jednotlivých zemích řeší databáze (Fabry registry a Fabry outcome survey), které shromažďují anonymní data pacientů s Fabryho chorobou z celého světa a jsou zdrojem velmi cenných poznatků o přirozeném průběhu nemoci a efektu léč-
by. Centrum pro Fabryho chorobu na II. interní klinice sleduje více než 100 pacientů a za použití ERT léčí 36 pacientů.
Randomizované, placebem kontrolované klinické studie s agalsidázou beta
Klinické studie s preparátem agalsidázou beta prokazují mimo regresi orgánového postižení a zlepšení kvality života také efekt na prognózu nemocných (pokles počtu závažných kardiovaskulárních a renálních příhod) [6].
Eng a kol. randomizovali 58 pacientů k léčbě α-galaktosidázou A (agalsidáza beta) v dávce 1 mg/kg 1krát za 14 dní a placebem [3]. Léčba byla podávána 20 týdnů a poté byli všichni pacienti léčeni v odslepené studii aktivní léčbou. Primárním cílovým ukazatelem bylo procento pacientů, u nichž došlo k vyčištění mikrovaskulárních endoteliálních depozit Gb3 v ledvinách. Hodnocena byla rovněž depozita v cévách myokardu a kůže. U 20 z 29 pacientů léčených agalsidázou beta po dobu 20 týdnů již nebyla přítomna žádná depozita v cévách ledvin oproti žádnému zlepšení u skupiny pacientů léčených placebem. Významně se rovněž snížilo procento pacientů s mikrovaskulárními depozity v myokardu a kůži. Mikrovaskulární depozita nebyla přítomna již téměř u žádného ze všech pacientů, jimž byla podávána aktivní léčba po 6 měsíců v rámci odslepené studie. Léčba byla dobře tolerována, pouze během podávání infuzí s aktivní léčbou se častěji vyskytovaly zimnice s třesavkou. Zpomalení infuze nebo premedikace zpravidla dostačovalo k odstranění této reakce.
Thurberg a kol. prokázali v radomizované, placebem kontrolované studii po 3 letech léčby agalsidázou beta vymizení kožních depozit (celkem 59 pacientů) [4].
Bierer a kol. prokázali v menší, rovněž randomizované, placebem kontrolované klinické studii zlepšení tolerance námahy po podávání agalsidázy beta [5].
Banikazemi a kol. sledovali 82 pacientů s mírnou nebo středně pokročilou renální insuficiencí. Střední délka sledování byla 18,5 měsíce, u nejdéle sledovaného pacienta byla délka sledování 35 měsíců. Nemocní byli randomizováni k léčbě agalsidázou beta v doporučené dávce 1 mg/kg 1krát za 14 dnů nebo k podávání placeba. Primárním cílovým ukazatelem byla první klinická příhoda (kardiální, renální nebo cerebrovaskulární). Ta se vyskytla u 42 % pacientů (z celkového počtu 31), kteří dostávali placebo, a pouze u 27 % pacientů (z celkového počtu 51) léčených agalsidázou beta. Poměr rizik činil 0,47 ve prospěch agalsidázy beta. Při analýze pacientů s glomerulární filtrací převyšující 55 ml/min byl efekt výraznější (poměr rizik 0,19, interval spolehlivosti 0,05–0,82) než u pacientů s funkcí nižší než 55 ml/min (relativní riziko 0,85). Podobně bylo lepších výsledků dosaženo u pacientů s malou proteinurií oproti pacientům se středně velkou nebo velkou proteinurií. Efekt léčby byl tedy podstatně lepší u pacientů s méně pokročilým postižením ledvin [6].
Zajímavá studie Lubandy a kol. prokázala příznivý efekt snížené dávky agalsidázy beta (0,3 mg/kg) následující po standardní dávce (1,0 mg/kg) na střádání ve tkáních [7]. Nepřítomnost střádání byla udržena u endotelií renálních kapilár v 90 % případů a u ostatních buněk ledvin a kůže v 70 %. Toto zjištění bylo důležité při řešení nedostatku agalsidázy beta kvůli výpadku ve výrobě a v některých zemích vedlo k povolení podávat redukované dávky léku.
Randomizované, placebem kontrolované klinické studie s agalsidázou alfa
Klinické studie s agalsidázou alfa prokazují zlepšení kvality života (ústup neuropatických bolestí, febrilií apod.) a ústup orgánového postižení (regrese hypertrofie levé komory a zlepšení histologických nálezů). Důležitou úlohu sehrála agalsidáza alfa v době nedostatku agalsidázy beta, kdy tento lék u řady pacientů nahradila, a dosavadní zkušenosti nesvědčí pro nižší účinnost léku či použité nižší dávky.
Schiffman a kol. randomizovali 26 pacientů k léčbě agalsidázou alfa nebo placebem v dávce 0,2 mg/kg 1krát za 14 dní a sledovali ovlivnění neuropatických bolestí [8]. Po 6 měsících aktivní léčby se signifikantně zmenšily bolesti nemocných a zlepšila se kvalita jejich života. V renálních biopsiích se při léčbě zmenšil počet glomerulů s rozšířením mesangia na podkladě depozit Gb3, pokles renálních funkcí byl signifikantně menší, došlo k 50% redukci plazmatické hladiny Gb3, zlepšilo se intraventrikulární vedení (měřeno šíří QRS komplexu) a vymizela blokáda pravého Tawarova raménka u jednoho pacienta. Léčba byla dobře tolerována, byly přítomny jen mírné reakce (třesavka), mizející po zpomalení infuze, po podání antihistaminik či nízkých dávek kortikoidů.
Moore a kol. randomizovali 26 pacientů k léčbě agalsidázou alfa nebo placebem v dávce 0,2 mg/kg 1krát za 14 dní a sledovali regionální prokrvení mozku pomocí pozitronové emisní tomografie po vizuální stimulaci a podání acetazolamidu [9]. Po 6měsíční léčbě došlo u pacientů dostávajících aktivní léčbu k normalizaci regionálního průtoku.
Hajioff a kol. vyšetřili 25 pacientů a zjistili, že u 36 % z nich byla přítomna bilaterální a u 40 % unilaterální ztráta sluchu ve vysokých frekvencích [10]. Randomizovali 15 pacientů se ztrátou sluchu k léčbě agalsidázou alfa nebo placebem v dávce 0,2 mg/kg podávané 1krát za 14 dní po dobu 6 měsíců a následně v otevřené studii trvající 36 měsíců. Po 6 měsících léčby se sluch u obou skupin dále signifikantně horšil. Ke zlepšení došlo teprve po 18 měsících a s délkou léčby se efekt léčby postupně zvyšoval. Uzavírali tedy, že ke zlepšení sluchu dochází postupně po delší době terapie.
Hughes a kol. studovali bezpečnost a účinnost léčby agalsidázou alfa u pacientů s postižením srdce [11]. Jednalo se o dvojitě slepou, placebem kontrolovanou studii u 15 dospělých mužů. Po 6 měsících léčby se významně snížila hmotnost levé komory měřená pomocí MRI (Magnetic Resonance Imaging) a pokleslo také množství Gb3 v myokardu o 20 % dle endomyokardiálních biopsií oproti jeho 10% nárůstu u pacientů léčených placebem. U aktivně léčených se také snížila hladina Gb3 v plazmě i moči. Léčba byla dobře snášena a nevyskytla se žádná závažná reakce, pouze u jednoho pacienta byla přítomna infuzní reakce, která se po premedikaci již vícekrát nevyskytla.
Hmotnost levé komory sledovali v retrospektivní studii také Kampmann a kol. [12]. Analyzovali výsledky 42 pacientů a srovnávali hmotnost levé komory (LK) měřenou echokardiograficky na začátku léčby, po 12 a 36 měsících léčby. U pacientů s hypertrofií levé komory došlo ke statisticky významné redukci hmotnosti po 12 a 36 měsících léčby. U pacientů bez hypertrofie LK došlo ke stabilizaci hmotnosti LK.
Nežádoucí účinky enzym-substituční léčby
Mezi nejčastější nežádoucí účinky patří infuzní reakce, které vznikají zpravidla na alergickém podkladě. Vyskytují se častěji než v 5 % případů. Nejčastěji se setkáváme s třesavkami, zimnicí, febriliemi, bolestmi hlavy či bolestmi kloubů a nevolností. Tyto reakce zpravidla odezní po zpomalení nebo přerušení infuze nebo podání antipyretik, antihistaminik či kortikoidů. Někdy se vyskytne kopřivka, svědění kůže, dušnost nebo otok hrtanu. Nejzávažnější reakcí je anafylaxe. Vyskytuje se ojediněle, ale s jejím rizikem je nutno vždy počítat.
V klinických studiích byla enzym-substituční léčba dobře snášena. Ve studiích Enga a kol. a Banikazemiho a kol. nebyly hlášeny žádné závažné reakce. Mírné infuzní reakce se vyskytovaly často, přibližně u 50 % pacientů. Klinické studie se však od běžné praxe mohou lišit zejména výběrem pacientů.
Dle našich zkušeností je léčba většinou dobře snášena. Infuzní reakce bývají mírné a ke snížení jejich výskytu často postačuje zpomalení rychlosti infuze. V několika případech si ale infuzní reakce vyžádaly záměnu jednoho preparátu za druhý.
Farmakologické chaperony
Rekombinantní α-galaktosidáza A má v neutrálním pH vnitřního prostředí krátký poločas a nízkou fyzikální stabilitu. Lze se domnívat, že zvýšení stability by mohlo výrazně ovlivnit účinnost léku. Tuto funkci mohou mít malé molekuly, kterým se říká chaperony. Vazbou na α-galaktosidázu A, ať už endogenního, nebo exogenního původu, prodlužují její poločas a potenciálně mohou výrazně zvýšit její účinnost. Při inkubaci fibroblastů od pacientů s Fabryho chorobou s molekulou označovanou jako AT1001 (1-deoxynojirimycin, DGJ) se čtyřnásobně zvýšilo vychytávání enzymu buňkami. Farmakologické chaperony fungují pouze u mutací měnících smysl kodonů („missens mutations“). Takových mutací je asi polovina.
V současné době probíhají klinické studie 3. fáze. První z nich je studie FACETS (The Fabry AT1001 Chaperone Efficacy, Therapeutics and Safety Study), která se týká pacientů bez enzym-substituční terapie [13]. Druhou je studie ATTRACT (AT1001 Therapy Compared to Enzyme Replacement in Fabry Patients with AT1001-responsive mutations: a Global Clinical Trial), která zahrnuje pacienty, jimž je stabilní dávka ERT podávána během alespoň 12 měsíců a poté jsou randomizováni k pokračování ERT nebo k podání chaperonu AT1001. Výsledky obou studií se očekávají v brzké době.
Genová terapie
Velkou naději pro pacienty s Fabryho chorobou představuje genová terapie. Probíhají slibné pokusy na zvířatech s podáváním retrovirů jako nosičů zdravého genu pro α-galaktosidázu A. Vycházejí ze skutečnosti, že i malé zvýšení aktivity enzymu (cca na 5 %) stačí zabránit orgánovému poškození. Problémem však zůstává imunitní reakce, která vede k poškození vektoru.
Nespecifická léčba
Příznivý vliv u chronických onemocnění ledvin s proteinurií mají inhibitory ACE a sartany. Proteinurie má zásadní prognostický význam také u Fabryho choroby, kde inhibitory ACE a sartany hrají důležitou úlohu nejen při kontrole hypertenze, ale také při kontrole proteinurie. Příznivý efekt inhibitorů ACE a sartanů byl extrapolován také na Fabryho chorobu. Experimentální zkušenosti tyto nálezy podporují, k dispozici však není žádná dvojitě slepá a placebem kontrolovaná klinická studie [14]. Tahir a kol. poukázali na fakt, že ERT samotná nedokázala kontrolovat proteinurii a že teprve po přidání inhibitorů ACE nebo sartanů došlo k její regresi [15]. Tento nález potvrdili v otevřené studii u vysoce rizikových pacientů, u kterých po přidání inhibitorů ACE nebo sartanů k léčbě poklesla proteinurie a stabilizovaly se renální funkce. Léčba byla dobře tolerována. Považujeme tedy za vhodné, pokud pacienti s proteinurií inhibitory ACE nebo sartany užívají.
Mnohé další projevy choroby vyžadují podpůrnou terapii. Při AV blokádách nebo jiných arytmiích dle potřeby implantujeme kardiostimulátory, defibrilátory, léčíme arytmie (farmakologicky, kardioverzí nebo intervenčně) a zvažujeme antikoagulační terapii. Při obstrukci ve výtokovém traktu levé komory provádíme septální ablaci. Při ischemické chorobě a angině pectoris podáváme antiagregancia a antianginózní preparáty (blokátory kalciového kanálu, betablokátory, nitráty), případně se provádí revaskularizace. Při selhání ledvin nahrazujeme jejich funkce dialýzou nebo transplantací ledvin, přinášejícími dobré výsledky [16]. K transplantaci se nepoužívají příbuzní dárci. Přežívání štěpu, a dokonce ani celková mortalita přesto není horší než u pacientů bez Fabryho choroby. Nemocní po transplantaci ledvin by měli být dále léčeni ERT. Štěp je sice chráněn vlastní produkcí enzymu, ale deficit enzymu v ostatních tkáních není transplantací korigován.
Časté jsou rovněž klasické rizikové faktory, které riziko kardiovaskulárních komplikací ještě zvyšují. Je proto nezbytné dodržovat zásady prevence kardiovaskulárních onemocnění a pacienty s Fabryho chorobou považovat za nemocné s vysokým rizikem. Ateroskleróza má u Fabryho choroby trochu jiný charakter. Zpravidla se setkáváme s difuzním postižením, které ztěžuje revaskularizaci.
Při gastrointestinálních symptomech, které se často podobají syndromu dráždivého tračníku a jejich příčina spočívá v poruše inervace trávicího traktu, je základem dieta s nižším obsahem tuků a častější příjem potravy v menších porcích. Dle potřeby podáváme prokinetika, pankreatické enzymy, inhibitory protonové pumpy nebo ondansetron. Farmakologická léčba má však omezenou účinnost.
Angiokeratomy, které vadí většinou z kosmetického hlediska, ale jež mohou také krvácet, lze ošetřit laserem nebo chirurgicky. Jejich tvorbu to ovšem nezastaví a příliš účinná není ani ERT. Rovněž syndrom karpálního tunelu, který se u pacientů s Fabryho chorobou vyskytuje častěji, lze řešit chirurgicky.
Problémem je léčba bolestí. Při chronických bolestech mohou být účinná antiepileptika (karbamazepin, fenytoin)
a gabapentin. Při bolestivých krizích používáme nesteroidní antirevmatika nebo opiáty. Důležité je pokusit se vystopovat a odstranit spouštěcí faktory, jako je stres, fyzická námaha, popř. vysoká nebo nízká teplota.
Významnou pomoc představuje psychologická podpora i užívání antidepresiv. Genetické poradenství je nezbytné při rodičovském plánování. Pomocí amniocentézy nebo odběru choriových klků můžeme vyšetřit pohlaví, aktivitu α-galaktosidázy A ve fetálních buňkách nebo stanovit mutaci.
Závěr
F abryho choroba je vzácné onemocnění ze střádání. V posledním desetiletí máme díky pokroku v oblasti lysozomálních nemocí ze střádání k dispozici specifickou enzym-substituční léčbu (tab. 1). Léčba se jeví jako účinná, ale především tehdy, začne-li se s ní včas. V blízké budoucnosti se můžeme setkat s další slibnou terapií malými molekulami – chaperony. Pravděpodobně ve vzdálenější budoucnosti budeme mít účinnou genovou léčbu. Nesmíme zapomínat na důležitou podpůrnou léčbu pacientů, podávání inhibitorů ACE a sartanů u pacientů s proteinurií, léčbu orgánových komplikací a psychologickou podporu. Předpokladem správné léčby je však vždy včasná diagnóza. Na Fabryho chorobu musíme myslet především při hypertrofii levé komory, nefropatii nebo postižení nervového systému nejasného původu. V centru Fabryho choroby jsou k dispozici lékaři i sestry na bezplatné telefonní lince (800 26 36 36), kteří jsou připraveni zodpovědět dotazy pacientů i zdravotníků, pomoci s diagnostikou, dispenzarizací i s léčbou. Fabryho choroba se vyznačuje multiorgánovým postižením, a vyžaduje proto multidisciplinární přístup. Spolupracujeme s pediatrickou, nefrologickou, neurologickou, dermatovenerologickou i ORL klinikou a zásadní je pro nás zejména spolupráce s Ústavem dědičných metabolických poruch 1. LF UK a VFN.
abryho choroba je vzácné onemocnění ze střádání. V posledním desetiletí máme díky pokroku v oblasti lysozomálních nemocí ze střádání k dispozici specifickou enzym-substituční léčbu (tab. 1). Léčba se jeví jako účinná, ale především tehdy, začne-li se s ní včas. V blízké budoucnosti se můžeme setkat s další slibnou terapií malými molekulami – chaperony. Pravděpodobně ve vzdálenější budoucnosti budeme mít účinnou genovou léčbu. Nesmíme zapomínat na důležitou podpůrnou léčbu pacientů, podávání inhibitorů ACE a sartanů u pacientů s proteinurií, léčbu orgánových komplikací a psychologickou podporu. Předpokladem správné léčby je však vždy včasná diagnóza. Na Fabryho chorobu musíme myslet především při hypertrofii levé komory, nefropatii nebo postižení nervového systému nejasného původu. V centru Fabryho choroby jsou k dispozici lékaři i sestry na bezplatné telefonní lince (800 26 36 36), kteří jsou připraveni zodpovědět dotazy pacientů i zdravotníků, pomoci s diagnostikou, dispenzarizací i s léčbou. Fabryho choroba se vyznačuje multiorgánovým postižením, a vyžaduje proto multidisciplinární přístup. Spolupracujeme s pediatrickou, nefrologickou, neurologickou, dermatovenerologickou i ORL klinikou a zásadní je pro nás zejména spolupráce s Ústavem dědičných metabolických poruch 1. LF UK a VFN.
Autoři by rádi připomněli obrovský přínos prof. MUDr. Milana Elledera, DrSc., v oblasti lysozomálních onemocnění ze střádání, vyslovili uznání jeho celoživotnímu dílu a současně vyjádřili zármutek nad náhlou ztrátou této vynikající osobnosti. Zároveň by autoři rádi ocenili zásluhy prof. MUDr. Jana Bultase, DrSc., a kolegů z II. interní kliniky kardiologie a angiologie VFN a 1. LF UK o založení Centra pro Fabryho chorobu a poděkovali za péči o pacienty trpící touto nemocí.
Seznam použité literatury
- [1] www.wiki.medpedia.com
- [2] Baehner F, Kampmann C, Whybra C. Enzyme replacement therapy in heterozygot females with Fabry disease: Results of a phase IIIB study. J Inherit Metab Dis 26; 2003; 26: 617–662.
- [3] Eng CM, Guffon N, Wilcox WR, et al. Safety and efficacy of recombinant human alpha-galactosidase A – replacement therapy in Fabry’s disease. N Engl J Med 2001; 345: 9–16.
- [4] Thurberg BL, Byers HR, Granter SR, et al. Monitoring the 3-year efficacy of enzyme replacement therapy in Fabry disease by repeated skin biopsies. J Invest Dermatol 2004; 122: 900–908.
- [5] Bierer G, Balfe D, Wilcox WR, Mosenifar Z. Improvement in serial cardiopulmonary exercise testing following enzyme replacement therapy in Fabry disease. J Inherit Metab Dis 2006; 29: 572–579.
- [6] Banikazemi M, Bultas J, Waldek S, et al. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med 2007; 146: 77–86.
- [7] Lubanda JC, Anijalg E, Bzdúch V, et al. Evaluation of a low dose, after a standard therapeutic dose, of agalsidase beta during enzyme replacement therapy in patients with Fabry disease. Genet Med 2009; 11: 256–264.
- [8] Schiffmann R, Kopp JB, Austin 3rd HA, et al. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA 2001; 285: 2743–2749.
- [9] Moore DF, Altarescu G, Herscovitch P, et al. Enzyme replacement reverses abnormal cerebrovascular responses in Fabry disease. BMC Neurol 2002; 2: 4.
- [10] Hajioff D, Goodwin S, Quiney R, et al. Hearing improvement in patients with Fabry disease treated with agalsidase alfa. Acta Paediatr Suppl 2003; 92: 28–30.
- [11] Hughes DA, Elliott PM, Shah J, et al. Effects of enzyme replacement therapy on the cardiomyopathy of Anderson-Fabry disease: a randomised, double-blind, placebo-controlled clinical trial of agalsidase alfa. Heart 2008; 94: 153–158.
- [12] Kampmann C, Linhart A, Devereux RB, Schiffmann R. Effect of agalsidase alfa replacement therapy on Fabry disease-related hypertrophic cardiomyopathy: a 12- to 36-month, retrospective, blinded echocardiographic pooled analysis. Clin Ther 2009; 31: 1966–1976.
- [13] The FACETS study. www.fabrystudy.com
- [14] Jain G, Warnock DG. Blood pressure, proteinuria and nephropathy in Fabry disease. Nephron Clin Pract 2011; 118: c43–8.
- [15] Tahir H, Jackson LL, Warnock DG. Antiproteinuric therapy and Fabry nephropathy: sustained reduction of proteinuria in patients receiving enzyme replacement therapy with agalsidase-beta. J Am Soc Nephrol 2007; 18: 2609–2617.
- [16] Ojo A, Meier-Kriesche HU, Friedman G, et al. Excellent outcome of renal transplantation in patients with Fabry’s disease. Transplantation 2000; 69: 2337–2339.
- [17] Wanner C. Fabry Disease Model: a rational approach to the management of Fabry disease. Clin Ther 2007; 29: supplement B.