Léčba pacientů s monogenním diabetem
Monogenní diabetes představuje zvláštní formu diabetu mellitu, jež je způsobena heterozygotním nosičstvím mutace v jednom genu a vyskytuje se u přibližně 2 % diabetických pacientů. Nejčastěji se setkáváme s glukokinázovým diabetem a HNF diabetem. Pacienti s glukokinázovým diabetem mají trvalou mírnou hyperglykemii, kterou ve většině případů není nutné léčit a která se vyskytuje v rodině v několika generacích. HNF diabetes je typický dědičností závažnějších forem diabetu, s velkým rizikem chronických diabetických komplikací a nutností farmakologické léčby. Léky první volby jsou deriváty sulfonylurey, ty mohou pomoci obejít genetický defekt v b-buňce a udržet pacienta dobře kompenzovaného a tím zpomalit rozvoj diabetických komplikací. Vzácně se setkáváme s pacienty s novorozeneckým diabetem a diabetem způsobeným mutací v genu pro inzulin. I v těchto případech lze optimalizovat léčbu podle výsledku genetického vyšetření.
Diabetes mellitus představuje heterogenní skupinu onemocnění s rozdílnou příčinou, ale s podobným průběhem. Oba nejběžnější typy diabetu – diabetes mellitus 1. typu (T1DM) a diabetes mellitus 2. typu (T2DM) – mají genetický základ, ale na jejich vzniku se podílejí odchylky ve více genech spolu s vlivy prostředí. Jedná se tedy o tzv. polygenní onemocnění. Přibližně 2 % diabetických pacientů trpí diabetem způsobeným odchylkou jen jediného genu, tedy monogenně podmíněným diabetem [1]. Většina případů monogenního diabetu je způsobena mutacemi v genech regulujících funkci β-buněk. Vzácně může diabetes vzniknout i díky mutaci vedoucí ve svém důsledku k velmi těžké inzulinové rezistenci nebo jako monogenně podmíněný autoimunitní diabetes mellitus. Velmi často je monogenní diabetes zaměněn za běžné formy diabetu (T1DM nebo T2DM). Přitom správná diagnóza onemocnění umožní výběr optimální terapie a terapeutické rozhodování je v mnohých aspektech odlišné od běžných doporučení pro většinové typy diabetu [2, 3].
 V klinické praxi se setkáváme především se dvěma hlavními skupinami pacientů s monogenním diabetem. Jsou to pacienti s glukokinázovým diabetem a HNF (hepatocytární nukleární faktor) diabetem. Na základě klinického podezření je v ideálním případě pacientovi a jeho rodině doporučeno molekulárně genetické vyšetření vzorku DNA, při němž se stanoví, v jakém genu je nesena mutace, a následně je určena přesná genetická diagnóza diabetu. Od této diagnózy se poté odvíjejí léčebná doporučení.
V klinické praxi se setkáváme především se dvěma hlavními skupinami pacientů s monogenním diabetem. Jsou to pacienti s glukokinázovým diabetem a HNF (hepatocytární nukleární faktor) diabetem. Na základě klinického podezření je v ideálním případě pacientovi a jeho rodině doporučeno molekulárně genetické vyšetření vzorku DNA, při němž se stanoví, v jakém genu je nesena mutace, a následně je určena přesná genetická diagnóza diabetu. Od této diagnózy se poté odvíjejí léčebná doporučení.
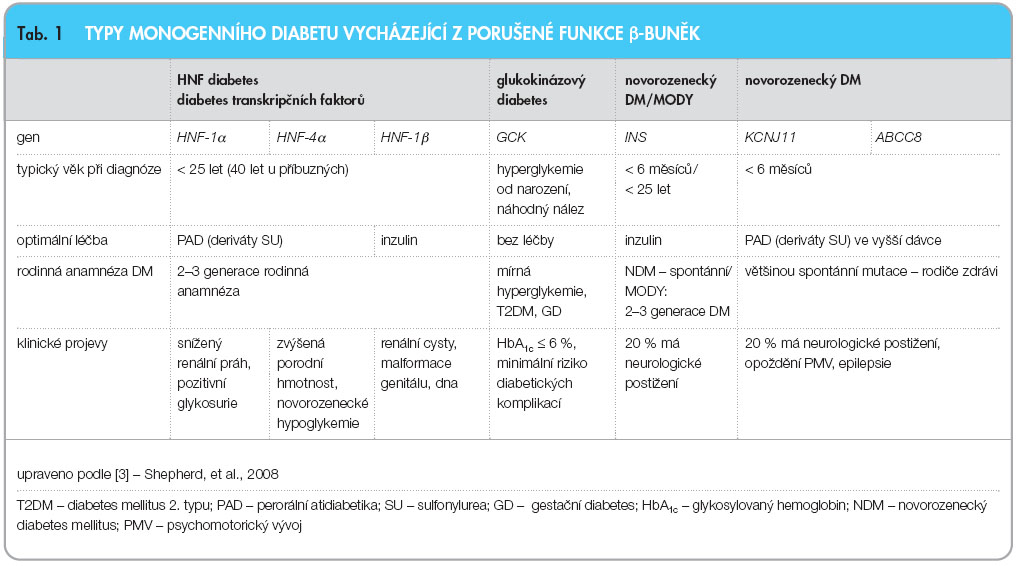
Nejčastější typy monogenního diabetu jsou shrnuty v tab. 1.
Glukokinázový diabetes (familiární mírná hyperglykemie, MODY2, GCK diabetes)
Pro glukokinázový diabetes způsobený heterozygotním nosičstvím mutace genu pro glukokinázu (GCK) je typická chronická mírná hyperglykemie od narození do stáří, s minimální progresí během života [1, 4]. Hyperglykemie při glukokinázovém diabetu vzniká na podkladě porušení funkce enzymu glukokinázy, který je senzorem β-buněk pro glukózu. V důsledku tohoto genetického defektu zahajují β-buňky sekreci inzulinu při vyšší glykemii, než je obvyklých 5 mmol/l. Tento defekt je přítomen již intrauterinně a zvýšenou glykemii nalačno lze zachytit v kterémkoli věku [2]. Osoby s mutací v genu pro glukokinázu jsou asymptomatické a ke zjištění poruchy dochází většinou náhodně. Věk při stanovení diagnózy závisí na prvním vyšetření glykemie a v průměru se pohybuje kolem 25 let (Ī 17) [2, 4]. Může se však jednat i o záchyt mírné hyperglykemie v novorozeneckém nebo kojeneckém věku. V České republice je glukokinázový diabetes nejčastěji diagnostikovanou formou monogenního diabetu [5, 6].
Následující projevy vedou k podezření na glukokinázový diabetes [7]:
- Hyperglykemie nalačno je stejná a stabilní během měsíců až let [8].
- HbA1c je typicky na horní hranici normy nebo ji lehce překračuje.
- Při oGTT (orální glukózový toleranční test) je vzestup glykemie mezi 0. a 120. minutou mírný (typicky o méně než 3 mmol/l), i když se doporučuje vzhledem k variabilitě v oGTT nepovažovat tuto hodnotu za absolutní pravidlo [8, 9].
- Rodiče mohou mít T2DM nebo být bez diabetu. Při testování má jeden z rodičů mírně zvýšenou glykemii nalačno v rozmezí 5,5–8,5 mmol/l, což potvrzuje autozomálně dominantní dědičnost [8].
Glukokinázový diabetes u dětí
U dětí představuje glukokinázový diabetes více než 50 % všech případů náhodně zachycené mírné hyperglykemie [10]. Jak již bylo zmíněno, může být zachycena v kterémkoli věku včetně novorozeneckého období. Ke standardnímu vyšetřovacímu programu patří rodinná anamnéza se stanovením glykemie nalačno u obou rodičů, dynamické zátěžové testy (oGTT, případně ivGTT – intravenózní glukózový toleranční test), stanovení hladiny autoprotilátek (anti-GAD, anti-IA2, případně také protilátek proti zinkovému transportéru a inzulinu) a HbA1c s cílem odlišit především premanifestní fázi T1DM. Na základě klinického podezření je pak doporučeno molekulárně genetické vyšetření, které potvrdí diagnózu monogenního diabetu. Děti s glukokinázovým diabetem jsou většinou sledovány v dětské diabetologické poradně s každoroční kontrolou glykemie, glykovaného hemoglobinu, hladiny lipidů a BMI. Žádná dietní ani léčebná opatření nejsou nutná [11]. Doporučuje se zdravý životní styl, prevence obezity a zamezení konzumace nápojů s velkým náhlým přívodem sacharidů (typu cola) [12]. V případě, že byla zahájena léčba malými dávkami inzulinu před stanovením genetické diagnózy, je možné ji bez náhrady vysadit. Pokud jsou tyto děti léčeny inzulinem, dochází k supresi vlastní tvorby inzulinu. To je patrně důvod, proč se u nich při léčbě inzulinem nevyskytují hypoglykemie [12].
Velmi vzácně může dojít k situaci, kdy mutace postihne obě alely glukokinázového genu. V tom případě se narodí dítě s těžkou formou permanentního novorozeneckého diabetu s nutností celoživotní léčby inzulinem [4]. V Čechách jsme takový případ zatím nezachytili.
Těhotenství pacientek s glukokinázovým diabetem je často prvním momentem, kdy se zjistí hyperglykemie. Díky aktivnímu screeningu glykemie je šance zachytit většinu těhotných žen s GCK diabetem, které po celé dětství a dospívání unikaly diagnóze. Mutace v genu pro GCK je nalezena cca u 1–4 % žen s gestačním diabetem [13]. Identifikace GCK diabetu mezi pacientkami s gestačním diabetem je důležitá, neboť tyto pacientky mají jiný průběh diabetu, a především nosičství mutace samo o sobě ovlivňuje velikost plodu. Ženy s GCK diabetem mají v těhotenství perzistující hyperglykemii nalačno, malý vzestup glykemie při oGTT (o méně než 3 mmol/l) a pozitivní rodinnou anamnézu [13]. Zvláště dědičný gestační diabetes v rodině může být nápadným vodítkem. Ženy s GCK diabetem jsou v průběhu těhotenství často léčeny inzulinem ke korekci hyperglykemie a ochraně plodu. Nicméně, jak bylo dokázáno, je to kombinace genotypu matky a dítěte, která rozhoduje o velikosti plodu, nikoli jen korekce glykemie [14]. Vlastní inzulin je pro vyvíjející se plod hlavním růstovým faktorem, jenž ovlivňuje velikost plodu. Také proto mají novorozenci diabetických matek s nekorigovanou hyperglykemií v těhotenství tendenci k makrosomii. Z toho vyplývá, že by děti matek s glukokinázovým diabetem měly být při narození makrosomické. Bylo skutečně dokázáno, že jinak zdravé děti, které se narodily matkám s GCK diabetem, jsou ve srovnání se svými příbuznými průměrně o 600 g těžší [15, 16]. Jiná situace ale nastává, jestliže dítě nese také mutaci v GCK genu. V tom případě má plod snížené vnímání mateřské hyperglykemie, nedochází k nadprodukci inzulinu u plodu, a nedojde tudíž ani k nadměrnému růstu plodu. Takové děti se pak rodí s normální porodní hmotností i přesto, že jejich matka měla v těhotenství neléčený gestační diabetes. Může také nastat situace, kdy zdravá normoglykemická matka nese plod s mutací v GCK genu. V takovém případě se dítě rodí s nižší porodní hmotností v průměru o 520 g. Stejná možnost může nastat také tehdy, pokud je matka v těhotenství léčena vysokými dávkami inzulinu s korekcí glykemie do normálních až nízkých hodnot. Dítě se pak může narodit s nízkou porodní hmotností [17]. Rozhodnutí o léčbě těhotné ženy s gestačním diabetem způsobeným mutací v GCK genu by tedy mělo být vždy učiněno na základě pečlivého sledování růstu plodu. Inzulin by se měl aplikovat především tehdy, hrozí-li dle ultrazvukového vyšetření makrosomie plodu. Pokud je inzulin aplikován, doporučují se plné dávky ke korekci glykemie. Po porodu je pak možné léčbu inzulinem ihned ukončit. Pacientka má pak trvalou mírnou hyperglykemii bez nutnosti další léčby.
Glukokinázový diabetes v dospělosti a stáří
Ve svém přirozeném průběhu představuje glukokinázový diabetes neprogredující poruchu metabolismu projevující se mírnou hyperglykemií až do stáří a je nejčastěji zaměňován za T2DM. Při oGTT se nejčastěji setkáváme s hraniční úrovní poruchy glukózové tolerance nebo těsně na hranici kritérií pro diabetes [8]. V této podobě zůstává průběh oGTT stabilní po mnoho let [9] s jen minimálním rizikem diabetických komplikací [16, 18, vlastní studie]. Pokud je pacient s přirozeným průběhem glukokinázového diabetu léčen, má glykemii nalačno i hladinu glykosylovaného hemoglobinu prakticky stejnou jako bez léčby. Nejlepším důkazem je situace, kdy po zjištění genetické diagnózy pacientovi léčbu zkusmo vysadíme a po několika měsících i po roce zjišťujeme zcela totožné výsledky, jaké měl pacient s využitím předchozí léčby (ať už se jednalo o malé dávky inzulinu, nebo perorální antidiabetika).
Na druhou stranu glukokinázový diabetes nepředstavuje žádnou ochranu před jinými typy diabetu, ale ani zvýšené riziko pro jejich vznik [19]. Obézní člověk s glukokinázovým diabetem proto velmi snadno může dospět do diabetu mellitu 2. typu s jeho typickými projevy (inzulinová rezistence a dyslipidemie se zvýšenou hladinou triglyceridů). Stejně tak u staršího pacienta, v jehož rodině se z jedné strany přenáší gkukokinázový diabetes a z druhé strany klasický diabetes 2. typu, může po mnoha letech neprogredující mírné hyperglykemie dojít ke zhoršení glykemií a k rozvoji diabetu mellitu 2. typu. V těchto případech se pacienti musí léčit dle doporučení pro léčbu pacientů s T2DM. Jejich cílová glykemie však bude stejná jako u glukokinázového diabetu, protože ani dietou, ani farmakoterapií se nesníží glykemie nalačno pod úroveň danou poruchou enzymu glukokinázy (s výjimkou plných substitučních dávek inzulinu, které zcela zablokují vlastní tvorbu) [7, 11].
Cílové hodnoty pro léčbu pacientů s glukokinázovým diabetem lze shrnout takto:
- glykemie nalačno: 5–8 mmol/l,
- glykosylovaný hemoglobin HbA1c: ≤ 6 % IFCC.
Ačkoli je zvláště hladina HbA1c nad úrovní doporučovanou ADA (Americká diabetologická asociace) i ČDS (Česká diabetologická společnost) pro zahájení intenzivní léčby diabetika, u pacientů s glukokinázovým diabetem se jedná o bezpečnou hranici nezvyšující riziko dlouhodobých diabetických komplikací. Je však třeba pacienty upozornit na zásady zdravého životního stylu a na nutnost udržet optimální tělesnou hmotnost s cílem předejít rozvoji diabetu mellitu 2. typu.
Metabolické známky přechodu glukokinázového diabetu do T2DM:
- glykemie nalačno > 8 mmol/l,
- glykosylovaný hemoglobin HbA1c > 6 %,
- dyslipidemie, zvýšená hladina triglyceridů,
- obezita.
HNF diabetes
HNF diabetes při užití moderní nomenklatury zahrnuje jednak HNF-1α diabetes (dříve MODY3 – Maturity Onset Diabetes of the Young) způsobený heterozygotní mutací v genu pro hepatocytární nukleární faktor 1a a dále HNF-4α diabetes (dříve MODY1), který vzniká na podkladě heterozygotního nosičství mutace v genu pro HNF-4α [1]. Vedle glukokinázového diabetu jsou to nejčastější formy monogenního diabetu, se kterými se můžeme v ordinaci diabetologa setkat. V České republice máme k 31. 12. 2010 potvrzenu genetickou diagnózu u 115 pacientů s HNF-1α diabetem a u 72 pacientů s HNF-4α diabetem (zdroj www.lmg.cz). Jejich počty v poslední době díky pečlivému výběru rodin s pozitivní rodinnou anamnézou stoupají každým dnem.
Klinickou charakteristiku pacientů s HNF diabetem lze shrnout takto [7]:
- Časně vznikající diabetes, který není životně závislý na inzulinu. Např. nevede ke ketoacidóze při vysazení inzulinu, může být dobře kompenzován malými dávkami inzulinu, má detekovatelný C-peptid při léčbě inzulinem při glykemii > 8 mmol/l mimo období iniciální remise (3 roky po manifestaci).
- Rodinná anamnéza diabetu. Diabetes může být léčen inzulinem a hodnocen jako T1DM, typicky je diagnostikován ve věku do 40 let, nejčastěji do 25 let života.
- Hodnoty zjištěné při oGTT v časném stadiu vykazují velký vzestup glykemie, a to vyšší než 5 mmol/l [7, 8]. Někteří pacienti mohou mít glykemii nalačno normální, a přitom ve 120. minutě se jasně projeví diabetická křivka [8].
- Chybění známek autoimunity (negativní autoprotilátky anti-GAD, anti-IA2, antiinzulinové protilátky).
- Pozitivní glykosurie při relativně normální glykemii způsobená sníženým renálním prahem pro glukózu (platí zvláště pro screening nediabetických příbuzných pacientů s HNF-1α diabetem) [8].
- Doložitelná senzitivita k derivátům sulfonylurey (SU) vedoucí až k hypoglykemii, navzdory špatné kompenzaci diabetu před zahájením léčby deriváty SU [20, 21].
- Navíc pouze u HNF-4α diabetu: tendence k novorozenecké makrosomii (přítomno u 56 % nositelů mutace), která může být provázena tranzientní novorozeneckou hypoglykemií (u 15 % nositelů mutace) [22].
Léčba pacientů s HNF diabetem
Léčba pacientů s HNF diabetem nejlépe dokumentuje absolutní důležitost spolupráce nemocného s lékařem a výběr správné terapie. Dobře spolupracující pacient může být dlouhodobě léčen perorálními antidiabetiky (deriváty SU) s malým rizikem rozvoje chronických diabetických komplikací. Na druhou stranu žádná jiná forma diabetu nepřináší tak zásadně rozdílné výsledky, pokud není u pacienta dostatečná adherence k léčbě. V takovém případě naopak dochází k extrémně rychlému rozvoji chronických komplikací diabetu se všemi důsledky, včetně výrazného zkrácení délky života. Nespolupracující pacient díky absenci ketoacidózy dokáže „fungovat“ s hodnotami glykemie výrazně překačujícími hranici 20 mmol/l (zaznamenali jsme případ dívky, jejíž průměrná denní glykemie byla 40 mmol/l při normálních parametrech acidobazické rovnováhy), což má zásadní dopad na rychlý rozvoj komplikací. Paradoxně i takový pacient může být úspěšně léčen perorálními antidiabetiky – jak dokazují např. kazuistiky [23, 24].
Pacienti s HNF diabetem mohou být iniciálně léčeni dietou [2]. Jedná se však o mladé osoby před klinickou manifestací diabetu – např. pokud byla genetická diagnóza stanovena u dětí a jsou pravidelně sledovány. Bylo prokázáno, že i dosud nediabetičtí nositelé mutace (děti a dospívající) mají ve srovnání se zdravými kontrolami sníženou tvorbu inzulinu (deficit β-buněk) a zvýšenou periferní inzulinovou senzitivitu. Nositelé mutace v HNF-1α genu mají navíc snížený renální práh pro glukózu [25, 26]. Glykosurie se u nich proto objevuje i po běžné zátěži glukózou a může sloužit jako screening HNF-1α diabetu u dětí a dospívajících.
V okamžiku vzestupu glykemií po jídle (např. při oGTT) nebo při zvýšení hladin HbA1c a celkovém zhoršení sledovaných diabetických parametrů je nutná již farmakologická léčba [8]. U mladých osob s geneticky potvrzenou diagnózou se jednoznačně doporučuje zahájit léčbu perorálními antidiabetiky (PAD), viz tab. 2. U osob iniciálně léčených inzulinem se následně doporučuje zvážit změnu léčby také na perorální antidiabetika. Hlavním cílem terapie je vybrat pro pacienta takovou možnost, která zajistí jeho motivaci k léčbě, spolupráci, a díky tomu nejlepší kompenzaci diabetu.
Již v roce 1997 bylo poprvé publikováno, že pacienti s HNF-1α diabetem mají dobrou senzitivitu (hypersenzitivitu) k podávání derivátů sulfonylurey. V roce 1998 byla publikována studie čítající 100 pacientů s HNF-1α diabetem, v níž třetina osob byla léčena pouze dietou, druhá třetina pacientů deriváty sulfonylurey a poslední třetina inzulinem [2]. Mladí pacienti s HNF-1α diabetem byli od stanovení diagnózy léčeni výhradně inzulinem na základě představy, že nejlepší kompenzace (s cílem oddálit chronické diabetické komplikace) je možné dosáhnout pouze intenzifikovaným inzulinovým režimem.
Pacienti s HNF diabetem jsou unikátní právě v tom, že léčbu PAD používanou přednostně u pacientů s T2DM lze často s lepším výsledkem použít u osob i dlouhodobě léčených inzulinem. Filozofie léčby vychází přímo z genetické podstaty onemocnění: mutace v genech HNF-1α a HNF-4α, jejichž proteiny fungují v jádře β-buňky jako transkripční faktory, zásadním způsobem ovlivní fungování β-buňky. Nejvíce je však postižena signalizační cesta. Je dokázáno, že funkce KATP kanálu (ATP senzitivní K+ kanál) nesoucího na sobě receptor pro deriváty sulfonylurey (SUR1) i následná část vedoucí k uvolnění inzulinu je zachována. Zdá se, že deriváty sulfonylurey mají schopnost obejít v b-buňce blok v mechanismu spouštění sekrece inzulinu způsobený vadným fungováním HNF-1α nebo HNF-4α v jádře. Aktivace kanálu KATP vnější cestou (podáním derivátu SU) umožní arteficiální depolarizaci membrány β-buňky vedoucí ve svém důsledku až ke spuštění sekrece inzulinu z inzulinových granul [20]. Dříve se hovořilo dokonce o jakési hypersenzitivitě k derivátům sulfonylurey prokázané na zvířecích modelech [27]. V humánní medicíně se však jedná spíše o zachovalou dobrou citlivost, neboť není rozdíl v jejich metabolismu mezi pacienty s HNF-1α diabetem a zdravými kontrolami [20, 28].
Dnes již existuje mnoho důkazů, že pacienti s HNF diabetem mají porušeno uvolňování inzulinu z β-buněk, zatímco citlivost k inzulinu je velmi dobrá [29]. Již Pearson a kol. v roce 2003 [20] popsali že pacienti s HNF diabetem mají pětkrát lepší reakci na gliklazid než na metformin, a významně se tak odlišují od pacientů s diabetem 2. typu. Sagen a kol. v roce 2005 prokázali, že pacienti s HNF-1α diabetem mají sníženou odpověď na glukózu při ivGTT, avšak po podání tolbutamidu se jejich odpověď výrazně zlepší [28]. Shepherdová v roce 2003 publikovala studii, ve které prokázala, že je možné úspěšně zahájit léčbu PAD u pacientů s HNF-1α diabetem i po mnoha letech inzulinoterapie (medián 20 let). Všichni pacienti byli schopni přejít na léčbu deriváty sulfonylurey bez rozvoje ketonurie nebo výrazné hyperglykemie. Pouze v jednom případě (muž léčený inzulinem 35 let) došlo ke zhoršení HbA1c a k návratu k léčbě inzulinem. V této studii byl využit gliklazid v průměrné dávce 80 mg denně (20–320 mg) [30].
V roce 2006 byla publikována studie porovnávající léčbu pacientů s HNF-1α diabetem pomocí glibenklamidu a nateglinidu [21]. Nateglinid a repaglinid představují moderní perorální antidiabetika, která se vyznačují rychlým nástupem účinku a jeho krátkým trváním. Ačkoli nepatří do skupiny derivátů sulfonylurey, mechanismus účinku je podobný – stimuluje KATP kanál β-buňky jinou cestou než deriváty SU. Hlavním cílem studie bylo posoudit vliv obou preparátů na vývoj postprandiální hyperglykemie a bezpečnost obou preparátů. Ukázalo se, že nateglinid lépe zabrání vzestupu glykemie po jídle a vykazuje nižší riziko hypoglykemií [21]. Tento preparát je ideální pro zahájení léčby zvyšující se glykemie u mladých pacientů s HNF diabetem (dospívající, případně děti) a u osob s nově manifestovaným diabetem, kde detekujeme především vzestup glykemie po zátěži. Ve zmíněné studii byl dospělým pacientům podáván nateglinid před jídlem v dávkování 30 mg/dávka [21].
Léčba dětských pacientů s HNF diabetem přinesla revoluci ve výběru preparátů. Před rozvojem poznatků o genetice diabetu byli dětští a dospívající pacienti léčeni výhradně inzulinem. PAD, dříve jednotně kontraindikovaná u pacientů ve věku do 18 let, zde mohou být efektivně a bezpečně využívána. Musíme však mít na paměti nutnost začínat velmi nízkými dávkami (čtvrtina zahajovací dávky u dospělých) a zvláště v počátcích léčby bedlivě monitorovat hypoglykemie [12]. Je možné využít celé spektrum derivátů sulfonylurey nebo repaglinid (viz tab. 2). 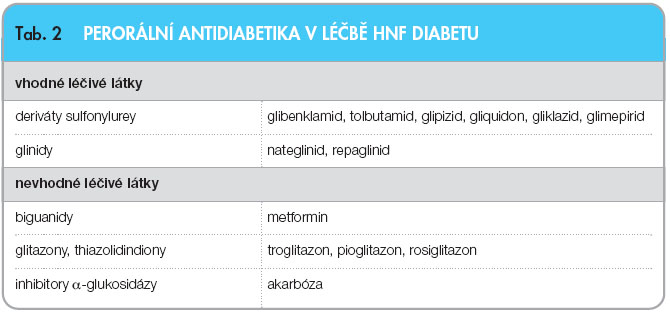 Právě repaglinid se výborně hodí k iniciální léčbě na pokrytí postprandiální hyperglykemie. Doporučuje se začít dávkou 0,5–2 mg před každým hlavním jídlem. V okamžiku, kdy repaglinid ke kontrole glykemie nestačí nebo se objevují ranní hyperglykemie, je možné zvolit některý z dlouhodoběji působících preparátů (např. gliklazid) podávaný 1–2krát denně [12].
Právě repaglinid se výborně hodí k iniciální léčbě na pokrytí postprandiální hyperglykemie. Doporučuje se začít dávkou 0,5–2 mg před každým hlavním jídlem. V okamžiku, kdy repaglinid ke kontrole glykemie nestačí nebo se objevují ranní hyperglykemie, je možné zvolit některý z dlouhodoběji působících preparátů (např. gliklazid) podávaný 1–2krát denně [12].
Určitou obavu při dlouhodobé léčbě deriváty sulfonylurey by mohla představovat s věkem se snižující sekrece inzulinu, a tedy nižší účinnost sulfonylureových derivátů. Nicméně stále častěji se objevují studie dokumentující dlouhodobou bezpečnost a účinnost terapie např. glibenklamidem (s naprostým vynecháním inzulinové léčby), která vedla také ke zlepšení spolupráce pacientů a zvýšení kvality jejich života [20, 30, 31]. V roce 2009 publikovala Shepherdová [32] práci, v níž bylo 79 % pacientů s diagnózou HNF-1α diabetu úspěšně převedeno z inzulinoterapie na léčbu PAD po stanovení genetické diagnózy. 71 % pacientů z původního počtu pokračovalo po 39 měsících (17–90 měsíců) stále v léčbě PAD s dobrou kompenzací diabetu. Důvody pro volbu inzulinoterapie byly plánování těhotenství anebo přání pacienta (předchozích mnoho let léčby inzulinem). Byly využity tyto látky: gliklazid, glimepirid, glibenklamid, glipizid. Nebyla zaznamenána žádná vážná hypoglykemie. Průměrná dávka glipizidu činila 1,3 mg/kg/ den (IQR 0,5–2,4 mg/kg/den). Maximální dávka před zahájením kombinované terapie s inzulinem byla 1,8–5,0 mg/kg/den a důvodem bylo pouze u pěti ze 43 osob zhoršení HbA1c [32].
Z přehledu publikovaných zkušeností vyplývá naléhavé doporučení vyzkoušet účinnost derivátů sulfonylurey u pacientů s HNF diabetem i u těch, kteří byli z různých důvodů (např. tíže symptomů při diagnóze, záměna za jiný typ diabetu) od počátku léčeni inzulinem [33]. Pro volbu této skupiny látek hovoří také významné zlepšení kvality života při léčbě deriváty sulfonylurey ve srovnání s inzulinoterapií [34]. Na druhou stranu u starších pacientů zvyklých dlouhou dobu na aplikaci inzulinu, při níž jsou dobře kompenzováni a přejí si u této léčby zůstat, není nutné za každou cenu léčbu měnit.
Pokud u starších, mnoho let léčených pacientů léčba deriváty sulfonylurey nevede k uspokojivé kompenzaci, je možné využít kombinovanou terapii s inzulinem [32], nebo dokonce s využitím inhibitorů dipeptidyl peptidázy 4 (gliptiny) [35].
Zásady léčby deriváty sulfonylurey u HNF diabetu lze shrnout takto:
- Deriváty suflonylurey mohou zlepšit kompenzaci diabetu a zahájení léčby deriváty SU má být indikováno v případě diabetiků špatně kompenzovaných dietou nebo inzulinem.
- Vzhledem k citlivosti k derivátům sulfonylurey hrozí hlavně na počátku léčby riziko hypoglykemie, proto je nutné opatrně upravovat dávku.
- Ukončení léčby deriváty SU má být pozvolné kvůli riziku metabolické dekompenzace [31].
Komplikace
Pacienti s HNF diabetem mají vysoké riziko mikrovaskulárních komplikací [36], které se zásadně odvíjí od kompenzace jejich diabetu dané správně zvolenou léčbou a především od compliance pacienta. Pacienti nedodržující léčebná doporučení mají vývoj komplikací akcelerován. Také riziko makrovaskulárních komplikací a z toho vyplývající mortality na kardiovaskulární onemocnění je vysoké [37]. Při porovnání diabetických nositelů mutace v HNF-1α genu a jejich příbuzných bez mutace byla zjištěna celkově vyšší mortalita v nižším věku (průměr ve studii 66 Ī 10 let). V 66 % případů bylo příčinou úmrtí právě kardiovaskulární onemocnění. V komplexní péči o starší pacienty s HNF diabetem je proto třeba sledovat také krevní tlak a hladinu lipidů, případně poruchy včas léčit dle běžných doporučení pro diabetické pacienty [37].
Novorozenecký diabetes
Diabetes diagnostikovaný do 6 měsíců věku dítěte nese souborný název novorozenecký diabetes a naprostá většina případů je monogenních. Může být permanentní (PNDM) nebo tranzietní diabetes (TNDM) a většina mutací je spontánních. Rodinná anamnéza diabetu je pozitivní jen v 15 % případů. Geny, jejichž mutace způsobují většinu případů novorozeneckého diabetu, jsou: KCNJ11 kódující Kir6.2 podjednotku KATP kanálu β-buňky, ABCC8 kódující SUR1 podjednotku KATP kanálu a genu pro inzulin (Ins) [38–40].
Převážná část pacientů s mutací v KCNJ11 a ABCC8 genu trpí izolovaným PNDM, u 20 % se mohou objevit sdružené neurologické symptomy. Nejzávažnějším, avšak naštěstí velmi vzácným projevem je nápadná porucha psychomotorického vývoje (PMV) s poruchou motorických a sociálních funkcí a generalizovanou epilepsií často s hypsarytmií v EEG [38]. Tato kombinace klinických projevů se označuje jako DEND syndrom (developmental delay, epilepsie a novorozenecký diabetes). Častěji se vyskytuje intermediální DEND syndrom, kdy pacienti mají méně závažnou poruchu PMV a netrpí epilepsií [39]. U všech pacientů se projevují známky inzulinové deficience. Ve 30 % případů se objeví ketoacidóza, většina z pacientů nemá detekovatelný C-peptid a jsou primárně léčeni inzulinem [39]. I přesto mohou být pacienti úspěšně léčeni PAD (deriváty SU) a při této léčbě mohou být lépe kompenzováni s nižším rizikem hypoglykemií. Potřebná dávka kalkulovaná na tělesnou hmotnost je v porovnání s dospělými vysoká. Pacienti většinou potřebují 0,5 mg/kg/glibenklamidu na den, v některých případech mohou vyžadovat až 1 mg/kg/den [39, 41]. Časem může dávka u některých pacientů klesat při zachování skvělé glykemické kontroly.
Diabetes způsobený mutací v genu pro inzulin
Tento typ diabetu může mít formu novorozeneckého diabetu [40], vzácně se také projevuje jako typický MODY diabetes, tj. s manifestací v průběhu dětství a dospívání s pozitivní rodinnou anamnézou diabetu ve 2–3 generacích. Diabetes způsobený mutací v genu pro inzulin je i při genetickém stanovení přesné diagnózy možné léčit pouze inzulinem [42]. U starších dospělých se někdy setkáváme s kombinací inzulinové léčby s perorálními antidiabetiky.
Molekulární diagnostika pacientů s klinickým podezřením na monogenní diabetes je v České republice dostupná v Laboratoři molekulární genetiky Pediatrické kliniky 2. lékařské fakulty Univerzity Karlovy a Fakultní nemocnice Motol (www.Lmg.cz) a je podporována finančním mechanismem Norského království CZ0100 a granty: IGA MZČR NT11402, MZ000064203.
Seznam použité literatury
- [1] Fajans SS, Bell GI, Polonsky KS. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med 2001; 345: 971–980.
- [2] Hattersley AT. Maturity-onset diabetes of the young: clinical heterogeneity explained by genetic heterogeneity. Diabet Med 1998; 15: 15–24.
- [3] Shepherd M. Our evolving understanding of monogenic diabetes: possibilities of improving glycaemic control following transfer from insulin to sulphonylureas. Prim Care Diabetes 2008; 2: 87–90.
- [4] Gloyn AL. Glucokinase (GCK) mutations in hyper- and hypoglycemia: maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemia in infancy. Hum Mutat 2003; 22: 353–362.
- [5] Pruhova S, Dusatkova P, Sumnik Z, et al. Glucokinase diabetes in 103 families from a country-based study in the Czech Republic: geographically restricted distribution of two prevalent GCK mutations. Pediatr Diabetes 2010; 11: 529–535.
- [6] Pruhova S, Ek J, Lebl J, et al. Genetic epidemiology of MODY in the Czech Republic: new mutations in the MODY genes HNF-4a, GCK and HNF-1a. Diabetologia 2003; 46: 291–295.
- [7] Ellard S, Bellanné-Chantelot C, Hattersley AT. Best practice guidelines for the molecular genetic diagnosis of maturity-onset diabetes of the young. Diabetologia 2008; 51: 546–553.
- [8] Stride A, Vaxillaire M, Tuomi T, et al. The genetic abnormality in the beta cell determines the response to an oral glucose load. Diabetologia 2002; 45: 427–435.
- [9] Martin D, Bellanné-Chantelot C, Deschamps I, et al. Long-term follow-up of oral glucose tolerance test-derived glucose tolerance and insulin secretion and insulin sensitivity indexes in subjects with glucokinase mutations (MODY2). Diabetes Care 2008; 31: 1321–1323.
- [10] Feigerlova E, Pruhova S, Dittertova L, et al. Aetiological heterogeneity of asymptomatic hyperglycaemia in children and adolescents. Eur J Pediatr 2006; 16: 446–452.
- [11] Hattersley AT. Molecular genetics goes to the diabetes clinic. Clin Med 2005; 5: 476-481.
- [12] Hattersley AT, Bruining J, Shield J, et al. ISPAD Clinical Practice Consensus Guidelines 2006–2007. The diagnosis and management of monogenic diabetes in children. Pediat Diabetes 2006; 7: 352–360.
- [13] Ellard S, Beards F, Allen LI, et al. A high prevalence of glucokinase mutations in gestational diabetic subjects selected by clinical criteria. Diabetologia 2000; 43: 250–253.
- [14] Hattersley AT, Beards F, Ballantyne E, et al. Mutations in the glucokinase gene of the fetus result in reduced birth weight. Nat Genet 1998; 19: 268–270.
- [15] Spyer G, Hattersley AT, Sykes JE, et al. Influence of maternal and fetal glucokinase mutations in gestational diabetes. Am J Obstet Gynecol 2001; 185: 240–241.
- [16] Velho G, Blanché H, Vaxillaire M, et al. Identification of 14 new glucokinase mutations and description of the clinical profile of 42 MODY-2 families. Diabetologia 1997; 40: 217–224.
- [17] Velho G, Hattersley AT, Froguel P. Maternal diabetes alters birth weight in glucokinase-deficient (MODY2) kindred but has no influence on adult weight, height, insulin secretion or insulin sensitivity. Diabetologia 2000, 43: 1060–1063.
- [18] Page RC, Hattersley AT, Levy JC, et al. Clinical characteristics of subjects with a missense mutation in glucokinase. Diabet Med 1995; 12: 209–217.
- [19] Murphy R, Ellard S, Hattersley AT. Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat Clin Pract Endocrinol Metab 2008; 4: 200–213.
- [20] Pearson ER, Starkey BJ, Powell RJ, et al. Genetic cause of hyperglycaemia and response to treatment in diabetes. Lancet 2003; 18: 1275–1281.
- [21] Tuomi T, Honkanen EH, Isomaa B, et al. Improved prandial glucose control with lower risk of hypoglycemia with nateglinide than with glibenclamide in patients with maturity-onset diabetes of the young type 3. Diabetes Care 2006; 29: 189–194.
- [22] Pearson ER, Boj SF, Steele AM, et al. Macrosomia and hyperinsulinaemic hypoglycaemia in patients with heterozygous mutations in the HNF4A gene. PLoS Med 2007; 4: e118.
- [23] Brunerová L, Trešlová L, Průhová Š, et al. Glibenklamid místo inzulinu: nová šance pro pacienty s diabetem MODY 3: (kazuistika). Vnitr Lek 2006; 52: 205–206.
- [24] Průhová Š, Dušátková P. Jak genetické vyšetření může pomoci ve výběru vhodné léčby diabetika. Lékařské listy ZDN 2010, 20: 3–5.
- [25] Stride A, Ellard S, Clark P, et al. Beta-cell dysfunction, insulin sensitivity, and glycosuria precede diabetes in hepatocyte nuclear factor-1alpha mutation carriers. Diabetes Care 2005; 28: 1751–1756.
- [26] Vaxillaire, M, Pueyo ME, Clément K, et al. Insulin secretion and insulin sensitivity in diabetic and non-diabetic subjects with hepatic nuclear factor-1alpha (maturity-onset diabetes of the young-3) mutations. Eur J Endocrinol 1999; 141: 609–618.
- [27] Boileau, P, Wolfrum, Ch, Shih, D, et al. Decreased glibenclamide uptake in hepatocytes of hepatocyte nuclear factor-1alpha-deficient mice: a mechanism for hypersensitivity to sulfonylurea therapy in patients with maturity-onset diabetes of the young, type 3 (MODY3). Diabetes 2002; 51: S343–S348.
- [28] Sagen JV, Pearson ER, Johansen A, et al. Preserved insulin response to tolbutamide in hepatocyte nuclear factor-1alpha mutation carriers. Diabet Med 2005; 22: 406–409.
- [29] Pearson ER, Pruhova S, Tack CJ, et al. Molecular genetics and phenotypic characteristics of MODY caused by hepatocyte nuclear factor 4alpha mutations in a large European collection. Diabetologia 2005; 48: 878–885.
- [30] Shepherd M, Pearson E, Houghton J, et al. No deterioration in glycemic control in HNF-1alpha maturity-onset diabetes of the young following transfer from long-term insulin to sulphonylureas. Diabetes Care 2003; 26: 3191–3192.
- [31] Pearson ER, Liddell WG, Shepherd M, et al. Sensitivity to sulphonylureas in patients with hepatocyte nuclear factor-1alpha gene mutations: evidence for pharmacogenetics in diabetes. Diabet Med 2000; 17: 543–545.
- [32] Shepherd M, Shields B, Ellard S, et al. A genetic diagnosis of HNF1A diabetes alters treatment and improves glycaemic control in the majority of insulin-treated patients. Diabet Med 2009; 26: 437–441.
- [33] Timsit J, Bellanné-Chantelot C, Dubois-Laforgue D, Velho G. Diagnosis and management of maturity-onset diabetes of the young. Treat Endocrinol 2005; 4: 9–18.
- [34] Shepherd M. Stopping insulin injections following genetic testing in diabetes: impact on identity. Diabet Med 2010; 27: 838–843.
- [35] Katra B, Klupa T, Skupien J, et al. Dipeptidyl peptidase-IV inhibitors are efficient adjunct therapy in HNF1A maturity-onset diabetes of the young patients-report of two cases. Diabetes Technol Ther 2010; 12: 313–316.
- [36] Isomaa B, Henricsson M, Lehto M, et al. Chronic diabetic complications in patients with MODY3 diabetes. Diabetologia 1998; 41: 467–473.
- [37] Steele AM, Shields BM, Shepherd M, et al. Increased all-cause and cardiovascular mortality in monogenic diabetes as a result of mutations in the HNF1A gene. Diabet Med 2010; 27: 157–161.
- [38] Gloyn AL, Pearson ER, Antcliff JF, et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med 2004; 350: 1838–1849.
- [39] Hattersley AT, Ashcroft FM. Activating mutations in Kir6.2 and neonatal diabetes: new clinical syndromes, new scientific insights, and new therapy. Diabetes 2005; 54: 2503–2513.
- [40] Edghill EL, Flanagan SE, Patch AM, et al. Neonatal Diabetes International Collaborative Group. Insulin mutation screening in 1,044 patients with diabetes: mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes 2008; 57: 1034–1042.
- [41] Pearson ER, Flechtner I, Nj/olstad PR, et al. Neonatal Diabetes International Collaborative Group. Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N Engl J Med 2006; 355: 467–477.
- [42] Boesgaard TW, Pruhova S, Andersson EA, et al. Further evidence that mutations in INS can be a rare cause od Maturity-Onset Diabetes of the Young (MODY). BMC Med Genet 2010; 11: 42.