Mechanismy prodloužení účinku inzulinu a jejich klinický význam
Používání inzulinu v klinické medicíně bylo od samého počátku spjato se snahou přizpůsobit léčbu inzulinem potřebám nemocných. V první polovině 20. století se metody prodloužení jeho účinku opíraly především o fyzikálně-chemické principy rozpustnosti proteinů a značný posun ve vývoji pak přineslo odhalení struktury inzulinového hexameru a využití principů genetického inženýrství.
Posun izoelektrického bodu kombinací molekul inzulinu s vysoce bazickými proteiny blíže k pH intersticiální tekutiny u inzulinu NPH způsobí tvorbu omezeně rozpustného precipitátu, v jehož formě zůstává i nadále v podkoží. Éra genetického inženýrství navázala na znalost struktury inzulinového hexameru racionálním designem inzulinových analog. V případě glarginu modifikace molekuly využívá ověřený princip posunu izoelektrického bodu a vede k tvorbě mikroprecipitátů inzulinu přímo v podkoží. V případě detemiru strategie kovalentní vazby kyseliny myristové na molekulu inzulinu využívá princip afinity hydrofobních partií molekuly inzulinu k molekulám albuminu. Cílem vývoje nové generace inzulinových analog je dosažení stabilního metabolického účinku v průběhu celých 24 hodin, jehož lze dosáhnout při splnění některých farmakologických parametrů (délka účinku inzulinu >> 24 hodin, nízká variabilita účinku inzulinu). Specifické vlastnosti inzulinu degludek jsou odvozeny od schopnosti tvorby multihexamerových komplexů a promítají se do vyrovnaného profilu jeho farmakodynamického účinku. Při délce biologického poločasu (t1/2) degludeku (průměrný t1/2 v rozmezí 24,4–26,8 hod.) se metabolické účinky jednotlivých dávek (při aplikaci 1krát denně) přirozeně překrývají, což napomáhá rovnoměrnému rozložení účinku v průběhu 24 hodin. Při vývoji inzulinu LY2605541 byl aplikován princip připojení molekuly polyethylenglykolu k molekule inzulinu lispro, což výrazně mění její „hydrodynamickou“ velikost. Tím je zpomaleno vstřebávání z podkoží i renální clearance tohoto inzulinu. LY2605541 vykazuje nízkou míru intraindividuální variability účinku a hepatoselektivní působení. Potřeby klinické praxe zaměřené na vyrovnaný metabolický účinek inzulinu, snížení rizika hypoglykemií a neutrální vliv na hmotnost léčených osob budou motivací k dalšímu vývoji inzulinových analog.
Úvod
Devadesát let používání inzulinu v klinické praxi vedlo nejen k záchraně mnoha životů, ale je zároveň spojeno s neutuchající snahou přizpůsobit léčbu inzulinem potřebám nemocných. Za zmíněné období byla zkoumána celá řada metod a vývoj se kvalitativně posunul zejména po odhalení struktury inzulinového hexameru a využitím principů genetického inženýrství. Výzkum na tomto poli zdaleka nekončí a sofistikované modifikace inzulinové molekuly rozšíří v budoucnosti terapeutické portfolio o další inzulinové přípravky. Nicméně obdiv si jistě zaslouží ti, kteří na samém počátku vývoje měli k dispozici z našeho pohledu pouze „primitivní“ technologie, a přesto využili tehdejší znalosti maximálním a naprosto fascinujícím způsobem.
Již záhy po zahájení léčby inzulinem v širším měřítku bylo zřejmé, že u řady osob dochází ke značnému kolísání glykemie během dne a příčinou těchto výkyvů hladin krevní glukózy byla „kinetika“ v té době jediného dostupného i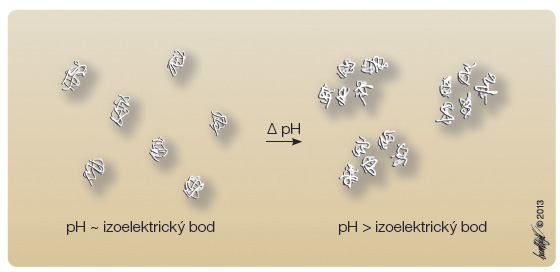 nzulinového přípravku, kterým byl inzulin hydrochlorid. Změny hladin glykemie po subkutánní aplikaci inzulinového přípravku svědčily pro jeho „relativně rychlou“ absorpci a účinek trvající přibližně 8 hodin. Potřeba změny „farmakokinetiky“ přípravků inzulinu byla formulována již roku 1928 dr. Joslinem: „Jak vděčni bychom byli, kdyby inzulin mohl být podáván ve formě, 1) jejíž účinek bude dvanáct a více hodin a 2) která nebude mít výrazný vrchol účinku díky rychlé absorpci, ale naopak pomalá absorpce bude pravidlem.“ [1]. Zřejmá potřeba klinické medicíny stimulovala již ve dvacátých letech minulého století vývoj metod zaměřených na zpomalení absorpce inzulinu z místa vpichu: 1) podáním inzulinu ve formě emulze v oleji či lecithinu, 2) aplikací inzulinu spolu s vazokonstrikčními látkami, 3) vývojem přípravků inzulinu omezeně rozpustných v intersticiální tekutině [2]. První dva zmíněné principy nenašly v praktické medicíně uplatnění, zatímco poslední varianta realizovaná Hagedornem byla úspěšná a opírala se o představu, že podání omezeně rozpustného precipitátu inzulinu vytvoří podkožní rezervoár, z něhož dojde k pozvolnější a prodloužené absorpci ve srovnání s inzulinem podávaným ve formě roztoku [3]. Vytvoření precipitátu inzulinu vycházelo teoreticky ze znalostí o rozpustnosti proteinů. Již na konci 19. století bylo známo, že proteiny jsou nejméně rozpustné v prostředí, jehož pH odpovídá jejich izoelektrickému bodu, v němž je celkový náboj proteinu nulový. Za této situace jsou odpudivé síly mezi dvěma molekulami proteinu nejmenší a ve vodném roztoku převáží síly přitažlivé. Tak dochází k agregaci proteinových molekul, a tedy k precipitaci proteinu, zatímco v prostředí s odlišným pH nese molekula proteinu různě silný náboj, který vede k elektrostatickému odpuzování jednotlivých molekul proteinu, viz obr. 1.
nzulinového přípravku, kterým byl inzulin hydrochlorid. Změny hladin glykemie po subkutánní aplikaci inzulinového přípravku svědčily pro jeho „relativně rychlou“ absorpci a účinek trvající přibližně 8 hodin. Potřeba změny „farmakokinetiky“ přípravků inzulinu byla formulována již roku 1928 dr. Joslinem: „Jak vděčni bychom byli, kdyby inzulin mohl být podáván ve formě, 1) jejíž účinek bude dvanáct a více hodin a 2) která nebude mít výrazný vrchol účinku díky rychlé absorpci, ale naopak pomalá absorpce bude pravidlem.“ [1]. Zřejmá potřeba klinické medicíny stimulovala již ve dvacátých letech minulého století vývoj metod zaměřených na zpomalení absorpce inzulinu z místa vpichu: 1) podáním inzulinu ve formě emulze v oleji či lecithinu, 2) aplikací inzulinu spolu s vazokonstrikčními látkami, 3) vývojem přípravků inzulinu omezeně rozpustných v intersticiální tekutině [2]. První dva zmíněné principy nenašly v praktické medicíně uplatnění, zatímco poslední varianta realizovaná Hagedornem byla úspěšná a opírala se o představu, že podání omezeně rozpustného precipitátu inzulinu vytvoří podkožní rezervoár, z něhož dojde k pozvolnější a prodloužené absorpci ve srovnání s inzulinem podávaným ve formě roztoku [3]. Vytvoření precipitátu inzulinu vycházelo teoreticky ze znalostí o rozpustnosti proteinů. Již na konci 19. století bylo známo, že proteiny jsou nejméně rozpustné v prostředí, jehož pH odpovídá jejich izoelektrickému bodu, v němž je celkový náboj proteinu nulový. Za této situace jsou odpudivé síly mezi dvěma molekulami proteinu nejmenší a ve vodném roztoku převáží síly přitažlivé. Tak dochází k agregaci proteinových molekul, a tedy k precipitaci proteinu, zatímco v prostředí s odlišným pH nese molekula proteinu různě silný náboj, který vede k elektrostatickému odpuzování jednotlivých molekul proteinu, viz obr. 1.
Inzulin Neutral Protamin Hagedorn (NPH) – téměř sedmdesát let na scéně
Samotný inzulin má izoelektrický bod přibližně 5,2, a proto v intersticiální tekutině s pH 7,4 zůstává ve formě roztoku a rychle se vstřebává. Realizace Hagedornovy úvahy o omezeně rozpustném precipitátu inzulinu proto vyžadovala kombinaci molekul inzulinu s vysoce bazickými proteiny, aby se izoelektrický bod výsledného komplexu přiblížil k pH inters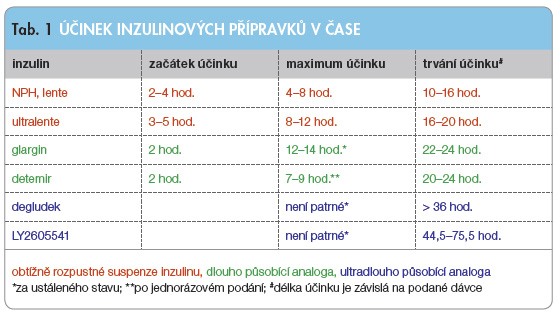 ticiální tekutiny. Jako bazické proteiny byly vybrány protaminy, polypeptidy izolované ze spermatu lososovitých ryb, bohaté na histidin, lysin a arginin. Při neutralizaci ve fosfátovém pufru na pH 7,3 byl právě protamin získaný ze pstruha duhového velmi účinný při tvorbě omezeně rozpustné suspenze [4]. Krátce po tomto objevu bylo zjištěno, že přidání zinku ke komplexu protamin-inzulin ještě výrazněji zpomalí absorpci inzulinu, a tak byl vyvinut přípravek nazývaný protamin-zinek-inzulin (PZI) [5]. Přidání zinku přispělo sice k prodloužení účinku až na 24 hodin, ale výrazná variabilita absorpce spojená s hypoglykemiemi a dále interakce s rychlým (regular) inzulinem při současném podání (tzn. ve stejné stříkačce tvořil nadbytečný protamin z PZI komplex s regular inzulinem) vyžadovaly řešení. O deset let později opět v Hagedornově laboratoři byl vyvinut další inzulinový přípravek, a to s využitím výsledků pozorování, že v přítomnosti zinku za fyziologického pH tvoří protamin a inzulin ve vyváženém množství krystaly protamin inzulinu. Tato krystalická forma inzulinu, protaminu a zinku se stala známou jako NPH nebo jako inzulin isophane [6]. Výhodami tohoto ppřípravku proti PZI byla nižší variabilita účinku a možnost míchání s rychle působícím inzulinem při zachování většiny farmakokinetických parametrů rychlého inzulinu, a proto zcela nahradil PZI. Jedná se nicméně o inzulin se středně dlouhým působením, a proto klinická praxe ukázala, že k adekvátní náhradě bazální sekrece je u osob s diabetem 1. typu nutné podávat tento inzulin 2krát denně (zejména pokud jsou podávána zároveň rychlá analoga inzulinu), viz tab. 1.
ticiální tekutiny. Jako bazické proteiny byly vybrány protaminy, polypeptidy izolované ze spermatu lososovitých ryb, bohaté na histidin, lysin a arginin. Při neutralizaci ve fosfátovém pufru na pH 7,3 byl právě protamin získaný ze pstruha duhového velmi účinný při tvorbě omezeně rozpustné suspenze [4]. Krátce po tomto objevu bylo zjištěno, že přidání zinku ke komplexu protamin-inzulin ještě výrazněji zpomalí absorpci inzulinu, a tak byl vyvinut přípravek nazývaný protamin-zinek-inzulin (PZI) [5]. Přidání zinku přispělo sice k prodloužení účinku až na 24 hodin, ale výrazná variabilita absorpce spojená s hypoglykemiemi a dále interakce s rychlým (regular) inzulinem při současném podání (tzn. ve stejné stříkačce tvořil nadbytečný protamin z PZI komplex s regular inzulinem) vyžadovaly řešení. O deset let později opět v Hagedornově laboratoři byl vyvinut další inzulinový přípravek, a to s využitím výsledků pozorování, že v přítomnosti zinku za fyziologického pH tvoří protamin a inzulin ve vyváženém množství krystaly protamin inzulinu. Tato krystalická forma inzulinu, protaminu a zinku se stala známou jako NPH nebo jako inzulin isophane [6]. Výhodami tohoto ppřípravku proti PZI byla nižší variabilita účinku a možnost míchání s rychle působícím inzulinem při zachování většiny farmakokinetických parametrů rychlého inzulinu, a proto zcela nahradil PZI. Jedná se nicméně o inzulin se středně dlouhým působením, a proto klinická praxe ukázala, že k adekvátní náhradě bazální sekrece je u osob s diabetem 1. typu nutné podávat tento inzulin 2krát denně (zejména pokud jsou podávána zároveň rychlá analoga inzulinu), viz tab. 1.
Úskalí využití zinku aneb rodina inzulinů „lente“
Dosažení úspěchu při zpomalení vstřebávání inzulinu z podkoží pomocí nekovalentní vazby s protaminem vedlo k dalšímu výzkumu zaměřenému na využití zinku nejen k eliminaci jiných proteinů, ale i k prodloužení absorpce z místa aplikace. Znalosti o působení vázaných či volných atomů zinku v krystalické matrix suspenze proteinu, kde napomáhají fyzikálně stabilizovat krystalickou strukturu a tak omezují rozpustnost dané bílkovinné suspenze, byly využity při vytvoření tzv. rodiny inzulinů „lente“ (semilente, lente, ultralente), tedy suspenzí inzulinu s různým množstvím zinku. Právě přebytek zinku se ukázal jako problematický pro inkompatibilitu při smíchání s přípravky rychle působícího inzulinu, ale především pro vysokou míru intraindividuální variability účinku a variability v závislosti na podané dávce [7].
Navíc se absorpce inzulinů ultralente lišila i dle místa aplikace [8, 9]. Podobně jako u inzulinu NPH byla přítomna variabilita účinku způsobená nedokonalou resuspenzí inzulinu před vlastní aplikací, která je považována za významný zdroj odlišného působení „stejné“ dávky mezi jednotlivými dny. Délka působení inzulinu ultralente byla sice delší než u inzulinu NPH, ale jen problematicky zajišťovala adekvátní pokrytí celého 24hodinového intervalu, viz tab. 1. Nicméně velmi výrazná intraindividuální variabilita účinku spojená s doloženým „dramatickým“ kolísáním glykemie léčených osob v nočních hodinách vedla k ukončení výroby této řady inzulinových přípravků [7]. Vývoj NPH inzulinu i inzulinů lente byl svého času jistě úspěchem a výsledkem maximálního využití tehdejších znalostí na poli chemie, a není proto překvapením, že tyto inzuliny i přes své nedostatky neměly po dobu několika dekád konkurenci.
Éra genetického inženýrství
Opravdoví rivalové se mohli objevit na scéně až po objevu krystalické struktury inzulinového hexameru, jehož analýza teprve umožnila racionální design inzulinových analog [10]. Technologie tvorby rekombinantní DNA pak poskytla klinické diabetologii jako první produkt krátkodobý inzulinový analog lispro. Právě využití krátkodobých analog v kombinaci s inzuliny NPH nebo ultralente zdůraznilo úskalí farmakokinetiky těchto bazálních inzulinů, protože h odnoty glykemií ráno nalačno a preprandiálně během dne byly vyšší než při použití kombinace s rychle působícím inzulinem, a tak v celkovém kontextu snižovaly potenciální výhody krátkodobých analog. Doplnění schématu léčby o druhou dávku inzulinu NPH (např. před polednem) se stalo u osob s diabetem 1. typu důležitým faktorem zlepšujícím celkovou efektivitu daného inzulinového režimu. Dalším významným stimulem hledání adekvátní náhrady bazální sekrece byly výsledky studie, v níž podávání inzulinu lispro v kontinuální infuzi vedlo k významnému poklesu hladin glykovaného hemoglobinu [11]. Tyto výsledky a úspěch krátkodobých inzulinových analog v omezení variability glykemií mezi jednotlivými dny a snížení frekvence hypoglykemií naznačovaly potřebu vývoje dlouho působících inzulinů s nižší intraindividuální variabilitou účinku.
odnoty glykemií ráno nalačno a preprandiálně během dne byly vyšší než při použití kombinace s rychle působícím inzulinem, a tak v celkovém kontextu snižovaly potenciální výhody krátkodobých analog. Doplnění schématu léčby o druhou dávku inzulinu NPH (např. před polednem) se stalo u osob s diabetem 1. typu důležitým faktorem zlepšujícím celkovou efektivitu daného inzulinového režimu. Dalším významným stimulem hledání adekvátní náhrady bazální sekrece byly výsledky studie, v níž podávání inzulinu lispro v kontinuální infuzi vedlo k významnému poklesu hladin glykovaného hemoglobinu [11]. Tyto výsledky a úspěch krátkodobých inzulinových analog v omezení variability glykemií mezi jednotlivými dny a snížení frekvence hypoglykemií naznačovaly potřebu vývoje dlouho působících inzulinů s nižší intraindividuální variabilitou účinku.
Tato výzva nezůstala bez odpovědi a biotechnologie umožnily realizaci několika odlišných strategií, viz tab. 2.
První z použitých strategií zpomalení absorpce inzulinu založená na posílení vazeb v hexameru inzulinu při použití kationtu kobaltu vytvořením komplexu Co3+-inzulin nevedla k úspěchu. Nicméně další strategie, která využ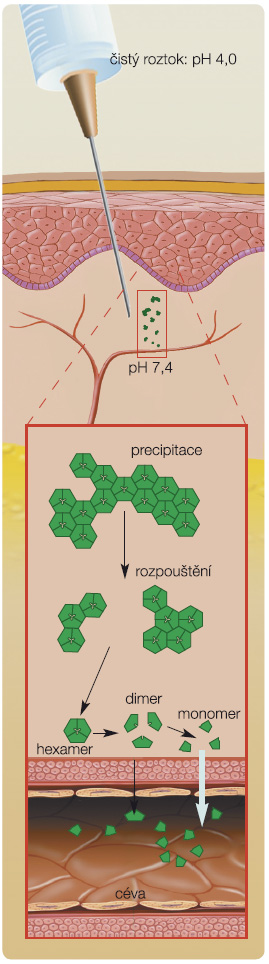 ila již ověřený princip posunu izoelektrického bodu molekuly inzulinu blíže k fyziologickému pH, vedla k vytvoření dvou modifikovaných molekul inzulinu. První byl Gly A21, Arg B27, Thr B30-NH2 humánní inzulin (NovoSol Basal), jehož klinické využití bylo ukončeno pro omezenou biologickou dostupnost snad způsobenou lokální zánětlivou reakcí v místě vpichu [12, 13]. Druhou byl Gly A21, Arg B31, Arg B32 humánní inzulin (inzulin glargin), u něhož posun izoelektrického bodu z 5,4 na 6,7 vede ke snížení rozpustnosti a k tvorbě mikroprecipitátů inzulinu v podkoží (obr. 2). Jeho produkce ve formě kyselého roztoku (pH 4,0) zajišťuje, že před podáním je inzulin glargin zcela rozpustný bez nutnosti resuspenze. Již úvodní srovnání inzulinu glargin s inzulinem ultralente svědčilo pro nižší variabilitu koncentrací inzulinu a glykemií při použití glarginu [14]. Podobně výsledky euglykemických clampů srovnávajících inzulin glargin s inzulinem NPH ukázaly zřetelné maximum účinku po 3–6 hodinách a odeznění účinku po 16 hodinách u inzulinu NPH, zatímco inzulin glargin měl maximum metabolického účinku (vyjádřené GIRmax, glucose infusion rate, rychlost infuze glukózy) zřetelně nižší v období 6–8 hodin po aplikaci a trvání jeho účinku v řadě případů přesáhlo 24 hodin [15, 16]. Na základě těchto výsledků byl inzulin glargin srovnáván s inzulinem NPH v klinických hodnoceních a jeho podávání v období před spaním vedlo k výraznějšímu poklesu hodnot glykemie nalačno, nižší variabilitě glykemií a k nižší frekvenci hypoglykemických epizod zejména nočních [17, 18].
ila již ověřený princip posunu izoelektrického bodu molekuly inzulinu blíže k fyziologickému pH, vedla k vytvoření dvou modifikovaných molekul inzulinu. První byl Gly A21, Arg B27, Thr B30-NH2 humánní inzulin (NovoSol Basal), jehož klinické využití bylo ukončeno pro omezenou biologickou dostupnost snad způsobenou lokální zánětlivou reakcí v místě vpichu [12, 13]. Druhou byl Gly A21, Arg B31, Arg B32 humánní inzulin (inzulin glargin), u něhož posun izoelektrického bodu z 5,4 na 6,7 vede ke snížení rozpustnosti a k tvorbě mikroprecipitátů inzulinu v podkoží (obr. 2). Jeho produkce ve formě kyselého roztoku (pH 4,0) zajišťuje, že před podáním je inzulin glargin zcela rozpustný bez nutnosti resuspenze. Již úvodní srovnání inzulinu glargin s inzulinem ultralente svědčilo pro nižší variabilitu koncentrací inzulinu a glykemií při použití glarginu [14]. Podobně výsledky euglykemických clampů srovnávajících inzulin glargin s inzulinem NPH ukázaly zřetelné maximum účinku po 3–6 hodinách a odeznění účinku po 16 hodinách u inzulinu NPH, zatímco inzulin glargin měl maximum metabolického účinku (vyjádřené GIRmax, glucose infusion rate, rychlost infuze glukózy) zřetelně nižší v období 6–8 hodin po aplikaci a trvání jeho účinku v řadě případů přesáhlo 24 hodin [15, 16]. Na základě těchto výsledků byl inzulin glargin srovnáván s inzulinem NPH v klinických hodnoceních a jeho podávání v období před spaním vedlo k výraznějšímu poklesu hodnot glykemie nalačno, nižší variabilitě glykemií a k nižší frekvenci hypoglykemických epizod zejména nočních [17, 18].
Výsledky studie monitorující vývoj hodnot glykemie v noci u podskupiny nemocných ukázaly, že při podávání inzulinu NPH dochází ke vzestupu glykemie kolem páté hodiny ranní. Naopak při podávání glarginu je tento vzestup glykemie v ranních hodinách potlačen [19].
Inzulin detemir, ε-lys B29-myristoyl des (B30) humánní inzulin
Jiná strategie vývoje dlouho působícího inzulinového analoga se opírá o kovalentní vazbu řetězce kyseliny myristové na ε-aminoskupinu lysinu v pozici B29. Tato modifikace podporuje reverzibilní vazbu molekuly inzulinu na albumin 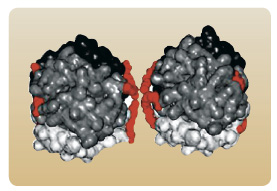 v místě aplikace a tak zpomaluje absorpci z podkožní tkáně. Odstranění threoninu v pozici B30 dále potencuje schopnost vazby inzulinového analoga na albumin. Připojením 14uhlíkatého řetězce kyseliny myristové se na povrchu molekuly inzulinu detemir vytváří hydrofobní oblast zajišťující vazbu na
v místě aplikace a tak zpomaluje absorpci z podkožní tkáně. Odstranění threoninu v pozici B30 dále potencuje schopnost vazby inzulinového analoga na albumin. Připojením 14uhlíkatého řetězce kyseliny myristové se na povrchu molekuly inzulinu detemir vytváří hydrofobní oblast zajišťující vazbu na 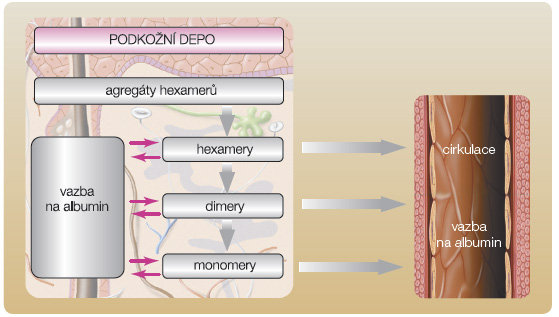 albumin a zároveň i agregaci molekul inzulinu. Inzulin detemir je před aplikací a krátce po aplikaci do podkoží přítomen ve formě hexamerů, ale jakmile dojde k náhradě adjuvancií (fenol a kresol) za fyziologické elektrolyty, dochází k tvorbě dihexamerů právě prostřednictvím hydrofobních oblastí na pólech hexamerů [20], viz obr. 3. Vytvoření dihexamerů se pravděpodobně významně podílí na opoždění disociace na monomery a spolu s vazbou na albumin zajišťuje zpomalení uvolňování detemiru do krevního řečiště [21], viz obr. 4.
albumin a zároveň i agregaci molekul inzulinu. Inzulin detemir je před aplikací a krátce po aplikaci do podkoží přítomen ve formě hexamerů, ale jakmile dojde k náhradě adjuvancií (fenol a kresol) za fyziologické elektrolyty, dochází k tvorbě dihexamerů právě prostřednictvím hydrofobních oblastí na pólech hexamerů [20], viz obr. 3. Vytvoření dihexamerů se pravděpodobně významně podílí na opoždění disociace na monomery a spolu s vazbou na albumin zajišťuje zpomalení uvolňování detemiru do krevního řečiště [21], viz obr. 4.
Po přesunu do cirkulace je inzulin detemir vázán v 98 % na albumin, ale tato vazba významněji neprodlužuje účinek inzulinu ve srovnání s vazbou na albumin v podkožním rezervoáru, neboť v cirkulaci je inzulin ve formě monomerů, a proto difunduje cévní stěnou do intersticiální tekutiny. Nicméně vazba detemiru na albumin v plazmě přináší jistý „pufrovací“ efekt, který omezuje farmakodynamickou variabilitu [22].
Tyto fyzikálně-chemické vlastnosti se příznivě promítly do jednoho z klíčových parametrů inzulinových přípravků, kterým je intraindividuální variabilita farmakodynamického účinku. Předpoklad byl ověřen v randomizované clampové studii, jejíž výsledky svědčily pro to, že inzulin detemir má vyšší míru konzistence profilů účinku v čase (koeficient variace, CV, 27 %) ve srovnání s inzulinem glargin (CV 48 %) a inzulinem NPH (CV 68 %); p < 0,0001 pro oba komparátory. Ve studii byla taktéž zjištěna nižší intraindividuální variabilita maximálního účinku detemiru (hodnocená GIRmax) vyjádřená koeficientem variace (CV 23 % pro inzulin detemir) ve srovnání s inzulinem NPH (CV 46 %; p < 0,0001) a inzulinem glargin (CV 36 %; p < 0,0001) [22].
Variabilita účinku jako klíčový parametr inzulinových přípravků
Tab. 1 shrnuje část základních farmakodynamických parametrů inzulinových přípravků (vyjádřených průměrem či rozmezím), ale pro úspěch léčby je často rozhodující druhá strana téže mince a tou je míra variability účinku.
Klinická zkušenost naznačuje, že při podkožní aplikaci inzulinu není často patrný shodný metabolický účinek, ač byla podána stejná dávka za srovnatelných podmínek. Rozdíly ve farmakodynamickém účinku jsou odvozeny od rozdílů v rychlosti absorpce dané nejen fyzikálně-chemickými vlastnostmi inzulinu, ale i fyziologickými faktory, jako je zvolené místo aplikace, průtok krve, kožní teplota, fyzická aktivita a stav hydratace. Intraindividuální variabilita účinku inzulinového přípravku má významný praktický dopad, neboť koreluje s rozdíly hypoglykemizujícího efektu při jednotlivých injekcích. U daného nemocného je tak intraindividuální variabilita účinku inzulinu spjata s mírou rizika vzniku jak hyper-, tak i hypoglykemie, které lze i odhadnout využitím nejnižšího a nejvyššího hypoglykemizujícího účinku inzulinu u daného nemocného [23]. Pro titraci dávky inzulinu by bylo optimální, pokud by stejná dávka inzulinu měla po každé aplikaci téměř identický metabolický účinek. Inzulinové přípravky s nízkou variabilitou účinku proto poskytují vyšší míru jistoty pro bezpečnou úpravu dávek, a tudíž mohou být vhodnějšími nástroji k dosažení cílů léčby.
Vlastnosti inzulinových přípravků, které ovlivňují intraindividuální variabilitu:
2.mikroprecipitáty vs. roztok inzulinu v místě aplikace
3.vazba na albumin – „nárazníkový efekt“ při náhlém výkyvu koncentrací,
4.délka účinku inzulinu (ve vztahu k překrývání metabolického účinku jednotlivých injekcí).
ad 1) Míra resuspenze a promíchání před vlastní aplikací je významným zdrojem variability účinku inzulinů dodávaných ve formě suspenze (t. č. již jen NPH) [24].
ad 2) V případě inzulinu glargin dochází po aplikaci k tvorbě mikroprecipitátů, které musí následně projít procesem opětovného rozpouštění a tyto procesy mohou být přirozeně proměnlivé. Pokud zůstává inzulinový přípravek v rozpustné formě i v místě vpichu (detemir, degludek), snižuje se míra variability uvolňování monomerů inzulinu do cirkulace [25].
ad 3) Vazba molekuly inzulinu (detemir, degludek) na albumin v plazmě vytváří „nárazníkový“ efekt, který omezuje kolísání volné frakce inzulinu, která je rozhodující pro metabolický účinek. Pokud se změní rychlost přesunu inzulinu z podkoží do cirkulace, téměř všechny molekuly se navážou na albumin, a proto jsou výsledné změny koncentrace volné frakce minimální.
ad 4) Inzuliny s délkou působení přesahující 24 hodin mají v rámci ustáleného stavu (odrážejícího klinickou situaci) přirozenou výhodu vedoucí ke snížení variability účinku tím, že celkový metabolický účinek velmi dlouho působícího analoga v určitém okamžiku je ovlivněn absorpcí inzulinu i z předchozích injekcí, a tak abnormální absorpce inzulinu z jednoho místa vpichu může být částečně kompenzována absorpcí z jiných míst [23].
Kde jsme a kam směřujeme?
Na základě euglykemických clampů považovaných za zlatý standard pro posouzení farmakodynamických profilů inzulinových přípravků lze konstatovat, že profily obou v současnosti používaných dlouho působících analog jsou charakterizovány mírným vzestupem a pozvolným poklesem hypoglykemizujícího účinku během 24 hodin. Obě analoga mají zřetelně delší působení a vyrovnanější profil účinku než inzulin NPH, ale nelze prozatím hovořit o kompletně „bezvrcholovém“ profilu účinku ideálního bazálního inzulinu [25].
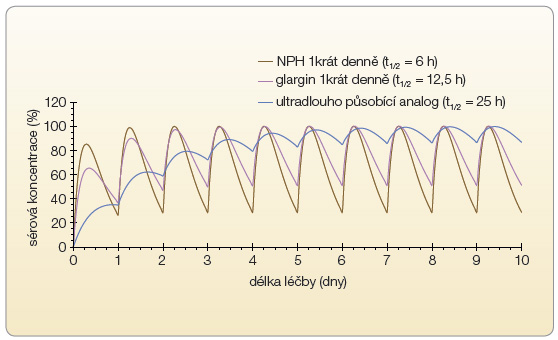 V praxi potřebný stabilní metabolický účinek inzulinového přípravku v průběhu celých 24 hodin vyžaduje splnění některých předpokladů daných obecnými farmakologickými pravidly. Aby se poměr maximálního a minimálního účinku inzulinu v průběhu 24 hodin blížil co nejvíce hodnotě 1, musí délka účinku inzulinu (za předpokladu jedné denní aplikace) značně přesahovat 24 hodin, viz graf 1. Dalším nezbytným kritériem je nízká variabilita účinku inzulinu, jíž lze dosáhnout především zachováním rozpustnosti inzulinu před aplikací i po podání do podkoží.
V praxi potřebný stabilní metabolický účinek inzulinového přípravku v průběhu celých 24 hodin vyžaduje splnění některých předpokladů daných obecnými farmakologickými pravidly. Aby se poměr maximálního a minimálního účinku inzulinu v průběhu 24 hodin blížil co nejvíce hodnotě 1, musí délka účinku inzulinu (za předpokladu jedné denní aplikace) značně přesahovat 24 hodin, viz graf 1. Dalším nezbytným kritériem je nízká variabilita účinku inzulinu, jíž lze dosáhnout především zachováním rozpustnosti inzulinu před aplikací i po podání do podkoží.
Nová generace dlouho působících inzulinových analog na obzoru
Vývoj nové generace inzulinových analog vychází opět z odlišných teoretických konceptů. V současné době prošel úspěšně klinickým zkoušením inzulinový přípravek schopný tvořit v podkoží multihexamerové komplexy (inzulin degludek) a III. fází klinického zkoušení prochází i bazální inzulin označený jako LY2605541, který je vytvořen připojením molekuly polyethylenglykolu (PEG) k molekule inzulinu lispro. Pro značné prodloužení farmakodynamického účinku těchto přípravků jsou někdy označovány jako ultradlouho působící analoga inzulinu.
Inzulin degludek
Dosažení výše uvedených farmakologických parametrů je v případě tohoto nového inzulinového přípravku opřeno o specifické fyzikálně-chemické vlastnosti, především o schopnost tvorby multihexamerových komplexů o vysoké molekulové hmotnosti, ovšem při zachování rozpustnosti těchto komplexů. Vývoji tohoto inzulinového analoga předcházel systematický výzkum podmínek, za nichž dochází k procesu řetězení hexamerů inzulinu. Tato situace nastane jen v případě, že je na molekulu inzulinu navázán ligand, který je schopen propojit hexamery inzulinu mezi ![Obr. 5 Změny konfigurace hexameru inzulinu za přítomnosti či nepřítomnosti fenolu; podle [44] – Derewenda, et al., 1989.](https://www.remedia.cz/photo-a-29168---.jpg) sebou, ale výsledek je značně závislý na konformaci hexamerů inzulinu, která se může za různých podmínek lišit. Inzulinové hexamery totiž mohou existovat v několika konformačních stavech podle toho, zda rezidua aminokyselin 1–6 v řetězci B inzulinového monomeru jsou v napjaté (T), nebo relaxované (R) konformaci [26], viz. obr. 5. Tyto změny polohy jsou závislé na vazbě molekul fenolu a chloridových aniontů, a mohou tak vést ke tvorbě tří různých konformací inzulinového hexameru: R6 je charakterizována uzavřením obou pólů hexameru, u R3T3 je jeden z pólů hexameru uzavřený a druhý otevřený a
sebou, ale výsledek je značně závislý na konformaci hexamerů inzulinu, která se může za různých podmínek lišit. Inzulinové hexamery totiž mohou existovat v několika konformačních stavech podle toho, zda rezidua aminokyselin 1–6 v řetězci B inzulinového monomeru jsou v napjaté (T), nebo relaxované (R) konformaci [26], viz. obr. 5. Tyto změny polohy jsou závislé na vazbě molekul fenolu a chloridových aniontů, a mohou tak vést ke tvorbě tří různých konformací inzulinového hexameru: R6 je charakterizována uzavřením obou pólů hexameru, u R3T3 je jeden z pólů hexameru uzavřený a druhý otevřený a![Obr. 6 Varianty konfigurace hexamerů inzulinu degludek; podle [28] – Jonassen, et al., 2012.](https://www.remedia.cz/photo-a-29169---.jpg) T6 má oba póly hexameru otevřeny (obr. 6). Význam různých konformací hexameru inzulinového analoga tkví v tom, že vazebná místa na další hexamery (atomy zinku nebo rezidua aminokyselin) jsou umístěna v jejich centru a mohou být buď exponována (při T konformaci), nebo chráněna (při R konformaci). Úvodní studie inzulinových analog acylovaných pomocí kyseliny cholové ukázaly vznik komplexů inzulinu o vysoké molekulové hmotnosti s prodlouženou délkou účinku [27]. Tyto výsledky vedly k hypotéze o možnosti vývoje inzulinových analog, u nichž boční řetězec „ušitý na míru“ zajistí tvorbu
T6 má oba póly hexameru otevřeny (obr. 6). Význam různých konformací hexameru inzulinového analoga tkví v tom, že vazebná místa na další hexamery (atomy zinku nebo rezidua aminokyselin) jsou umístěna v jejich centru a mohou být buď exponována (při T konformaci), nebo chráněna (při R konformaci). Úvodní studie inzulinových analog acylovaných pomocí kyseliny cholové ukázaly vznik komplexů inzulinu o vysoké molekulové hmotnosti s prodlouženou délkou účinku [27]. Tyto výsledky vedly k hypotéze o možnosti vývoje inzulinových analog, u nichž boční řetězec „ušitý na míru“ zajistí tvorbu 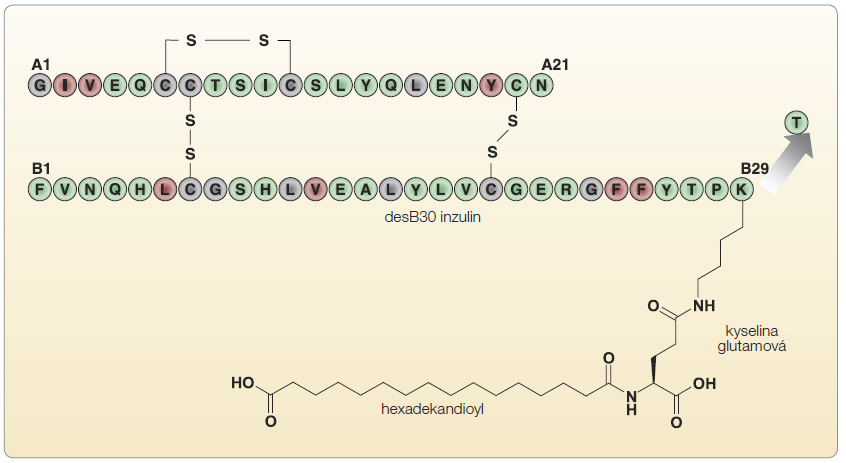 multihexamerů inzulinu, a tím výrazné prodloužení účinku inzulinového analoga. Na základě této představy vznikla inzulinová analoga vytvořená acylací ε-aminoskupiny lysinu v pozici B29 biosyntetického desB30 humánního inzulinu. Jako ligandy byly použity mastné kyseliny, ale na rozdíl od konstrukce molekuly detemiru byla navíc využita kyselina glutamová jako „vložka“ mezi molekulu inzulinu a mastné kyseliny (obr. 7). Z řady testovaných molekul měl nejpříznivější fyzikálně-chemické vlastnosti inzulinový analog acylovaný hexadekandioyl-gamma-L-glutamátem (inzulin degludek), který byl schopen v podkoží tvořit multihexamery o molekulové hmotnosti větší než 5 MDa [28]. U tohoto inzulinového analoga výzkum ukázal změny konformace hexamerů inzulinu podle chemického složení okolí, a tak např. v přítomnosti molekul fenolu ve farmaceutickém přípravku je inzulin degludek v konformaci R3T3, která umožní tvorbu dihexamerů pomocí vazby dvou T3 pólů k sobě
multihexamerů inzulinu, a tím výrazné prodloužení účinku inzulinového analoga. Na základě této představy vznikla inzulinová analoga vytvořená acylací ε-aminoskupiny lysinu v pozici B29 biosyntetického desB30 humánního inzulinu. Jako ligandy byly použity mastné kyseliny, ale na rozdíl od konstrukce molekuly detemiru byla navíc využita kyselina glutamová jako „vložka“ mezi molekulu inzulinu a mastné kyseliny (obr. 7). Z řady testovaných molekul měl nejpříznivější fyzikálně-chemické vlastnosti inzulinový analog acylovaný hexadekandioyl-gamma-L-glutamátem (inzulin degludek), který byl schopen v podkoží tvořit multihexamery o molekulové hmotnosti větší než 5 MDa [28]. U tohoto inzulinového analoga výzkum ukázal změny konformace hexamerů inzulinu podle chemického složení okolí, a tak např. v přítomnosti molekul fenolu ve farmaceutickém přípravku je inzulin degludek v konformaci R3T3, která umožní tvorbu dihexamerů pomocí vazby dvou T3 pólů k sobě ![Obr. 8 Přesmyk konfigurací hexameru inzulinu degludek umožňuje spontánní řetězení hexamerů v místě vpichu; podle [28] – Jonassen, et al., 2012.](https://www.remedia.cz/photo-a-29171---.jpg) navzájem (obr. 8). Mimořádný význam pro farmakokinetické vlastnosti degludeku má právě konverze z R3T3 hexameru na T6 konformaci hexameru po disociaci molekul fenolu v místě aplikace inzulinu. Tak je navozeno otevření hexameru inzulinu na obou pólech a mastné kyseliny mohou vytvořit vazbu na atom zinku sousedního hexameru z obou stran, což umožní tvorbu řetězce hexamerů řádově se stovkami hexamerových jednotek. Následná postupná disociace atomů zinku vede k tomu, že se koncový hexamer řetězce rozpadne na dimery a monomery, které snadno projdou do krevního řečiště. Tento proces pozvolné disociace koncových hexamerů vede k postupnému uvolňování inzulinu do cirkulace a plynulé degradaci řetězce hexamerů, což se příznivě odráží v řadě farmakokinetických parametrů [29]. Tvorba multihexamerů v místě vpichu je novým fenoménem a vyžaduje zhodnocení z hlediska možné imunologické odpovědi nebo lokálních reakcí. Dosavadní data neprokazují zvýšenou tvorbu specifických protilátek proti degludeku a podobně i kožní reakce v místě vpichu se objevovaly jen vzácně a bez známek vyšší incidence proti komparátoru, jímž byl inzulin glargin [30].
navzájem (obr. 8). Mimořádný význam pro farmakokinetické vlastnosti degludeku má právě konverze z R3T3 hexameru na T6 konformaci hexameru po disociaci molekul fenolu v místě aplikace inzulinu. Tak je navozeno otevření hexameru inzulinu na obou pólech a mastné kyseliny mohou vytvořit vazbu na atom zinku sousedního hexameru z obou stran, což umožní tvorbu řetězce hexamerů řádově se stovkami hexamerových jednotek. Následná postupná disociace atomů zinku vede k tomu, že se koncový hexamer řetězce rozpadne na dimery a monomery, které snadno projdou do krevního řečiště. Tento proces pozvolné disociace koncových hexamerů vede k postupnému uvolňování inzulinu do cirkulace a plynulé degradaci řetězce hexamerů, což se příznivě odráží v řadě farmakokinetických parametrů [29]. Tvorba multihexamerů v místě vpichu je novým fenoménem a vyžaduje zhodnocení z hlediska možné imunologické odpovědi nebo lokálních reakcí. Dosavadní data neprokazují zvýšenou tvorbu specifických protilátek proti degludeku a podobně i kožní reakce v místě vpichu se objevovaly jen vzácně a bez známek vyšší incidence proti komparátoru, jímž byl inzulin glargin [30].
![Graf 2 Průměrné profily GIR (glucose infusion rate, rychlost infuze glukózy) inzulinu degludek v průběhu 24 hodin za ustáleného stavu u osob s diabetem 2. typu; podle [32] – Heise, et al., 2012.](https://www.remedia.cz/photo-a-29172---.jpg) Vzhledem k přítomnosti mastné kyseliny na každé molekule inzulinu je podobně jako u detemiru zachována afinita k albuminu, která dále tlumí variabilitu koncentrací volných frakcí inzulinu. Výše uvedené fyzikálně-chemické vlastnosti inzulinu se promítají do farmakokinetických a farmakodynamických parametrů tohoto inzulinového analoga. Při použití euglykemického glukózového clampu bylo zjištěno, že metabolický efekt degludeku přesahuje 42 hodin (déle již lačnění probandů s diabetem 1. typu nebylo z etických důvodů realizováno) [31]. Výsledky další „clampové“ studie u osob s diabetem 2. typu posuzující metabolický efekt několika dávek degludeku (0,4 U/kg; 0,6 U/kg; 0,8 U/kg) za rovnovážného stavu pomocí parametru GIR svědčí pro stabilní ploché
Vzhledem k přítomnosti mastné kyseliny na každé molekule inzulinu je podobně jako u detemiru zachována afinita k albuminu, která dále tlumí variabilitu koncentrací volných frakcí inzulinu. Výše uvedené fyzikálně-chemické vlastnosti inzulinu se promítají do farmakokinetických a farmakodynamických parametrů tohoto inzulinového analoga. Při použití euglykemického glukózového clampu bylo zjištěno, že metabolický efekt degludeku přesahuje 42 hodin (déle již lačnění probandů s diabetem 1. typu nebylo z etických důvodů realizováno) [31]. Výsledky další „clampové“ studie u osob s diabetem 2. typu posuzující metabolický efekt několika dávek degludeku (0,4 U/kg; 0,6 U/kg; 0,8 U/kg) za rovnovážného stavu pomocí parametru GIR svědčí pro stabilní ploché ![Graf 3 Farmakodynamický profil inzulinu degludek za ustáleného stavu; podle [32] – Heise, et al., 2012. AUC – plocha pod křivkou plazmatických koncentrací; GIR – glucose infusion rate, rychlost infuze glukózy](https://www.remedia.cz/photo-a-29173---.jpg) profily účinku tohoto inzulinu během celého 24hodinového intervalu (graf 2). Tento fakt také vyjadřuje poměr metabolického účinku v intervalu 0–12 hodin vs. 12–24 hodin po aplikaci, který byl velmi blízký hodnotě 50 : 50 (graf 3). Obdobné výsledky rovnoměrného účinku platily i při rozdělení na 6hodinové intervaly. Podobně i zde farmakodynamický účinek degludeku přesáhl u všech vyšetřovaných osob délku trvání clampu, tj. 26 hodin [32].
profily účinku tohoto inzulinu během celého 24hodinového intervalu (graf 2). Tento fakt také vyjadřuje poměr metabolického účinku v intervalu 0–12 hodin vs. 12–24 hodin po aplikaci, který byl velmi blízký hodnotě 50 : 50 (graf 3). Obdobné výsledky rovnoměrného účinku platily i při rozdělení na 6hodinové intervaly. Podobně i zde farmakodynamický účinek degludeku přesáhl u všech vyšetřovaných osob délku trvání clampu, tj. 26 hodin [32].
Farmakokinetika léčiva s dlouhým poločasem, jakým je inzulin degludek, je charakterizována v prvních dnech léčby rostoucími údolními koncentracemi (v čase těsně před podáním další dávky) a ustáleného stavu je dosaženo po zhruba třech dnech léčby. Pro inzulin degludek dále platí, že expozice inzulinu, tj. plocha pod křivkou plazmatických koncentrací v rámci jednoho dávkovacího intervalu, a maximální plazmatické koncentrace stoupají úměrně podané dávce inzulinu. Podobně jako u farmakodynamického účinku byla expozice inzulinu rovnoměrně rozložena v průběhu 24hodinového dávkovacího intervalu. Hodnocení farmakokinetických profilů v průběhu 120 hodin po poslední podané dávce ukázalo, že plazmatické hladiny degludeku postupně klesaly a byly detekovatelné i 120 hodin po poslední dávce u všech sledovaných subjektů při všech testovaných dávkách. Pro 3 uvedené dávky degludeku se pohyboval průměrný biologický poločas (t1/2) v rozmezí 24,4–26,8 hodiny a terminální poločas průřezově pro testované dávky byl odhadnut na 25,1 hodiny [32].
![Graf 4 Srovnání parametrů dávkovacích schémat dlouhodobě působících inzulinových analog; podle [45] – DeVries, et al., 2007.](https://www.remedia.cz/photo-a-29174---.jpg) Právě délka poločasu eliminace degludeku se podílí na vyrovnaném profilu farmakodynamického účinku tohoto inzulinu. Při této délce poločasu inzulinového analoga se metabolické účinky jednotlivých injekcí (i při aplikaci 1krát denně) přirozeně překrývají, což napomáhá rovnoměrnému rozložení účinku v průběhu 24 hodin bez patrného vrcholu a připívá k omezení variability účinku (graf 1). Naopak u současných inzulinových přípravků působících přibližně 24 hodin či méně platí (při aplikaci 1krát denně), že koncentrace inzulinu začíná v podstatě z nuly, stoupá k vrcholovým hladinám (cmax) a poté opět klesá k údolní hodnotě (cmin). Toto kolísání hladin inzulinu (a odpovídající kolísání metabolického účinku) lze redukovat častější aplikací, např. 2krát denně, protože za této situace účinek následující dávky překrývá pokles účinku dávky předchozí [33], viz graf 4.
Právě délka poločasu eliminace degludeku se podílí na vyrovnaném profilu farmakodynamického účinku tohoto inzulinu. Při této délce poločasu inzulinového analoga se metabolické účinky jednotlivých injekcí (i při aplikaci 1krát denně) přirozeně překrývají, což napomáhá rovnoměrnému rozložení účinku v průběhu 24 hodin bez patrného vrcholu a připívá k omezení variability účinku (graf 1). Naopak u současných inzulinových přípravků působících přibližně 24 hodin či méně platí (při aplikaci 1krát denně), že koncentrace inzulinu začíná v podstatě z nuly, stoupá k vrcholovým hladinám (cmax) a poté opět klesá k údolní hodnotě (cmin). Toto kolísání hladin inzulinu (a odpovídající kolísání metabolického účinku) lze redukovat častější aplikací, např. 2krát denně, protože za této situace účinek následující dávky překrývá pokles účinku dávky předchozí [33], viz graf 4.
Tyto teoretické předpoklady potvrdily výsledky clampové studie u osob s diabetem 1. typu srovnávající intraindividuální variabilitu farmakodynamického účinku inzulinů degludek a glargin. Intraindividuální variabilita celkového metabolického efektu vyjádřeného plochou pod křivkou v ustáleném stavu AUCGIR,0-24h byla výrazně nižší při použití degludeku (CV 20 %) ve srovnání s glarginem (CV 82 %), p < 0,0001. Podobně i variabilita ![Graf 5 Intraindividuální variabilita účinku inzulinu degludek ve srovnání s inzulinem glargin; podle [23] – Heise, et al., 2012. CV – koeficient variability; AUC – plocha pod křivkou plazmatických koncentrací; GIR – glucose infusion rate, rychlost infuze glukózy](https://www.remedia.cz/photo-a-29175---.jpg) maximálního metabolického efektu GIRmax byla signifikantně nižší u degludeku (CV 18 %) ve srovnání s glarginem (CV 60 %), p < 0,0001, viz graf 5 [23]. I když je vždy potřebná značná opatrnost, pokud se data z experimentální situace přenášejí do reálné léčebné praxe, tak v tomto případě se předpokládané snížení rizika vzniku hypoglykemií při použití degludeku potvrdilo výsledky klinických studií. Opět ve srovnání s inzulinem glargin, při dosažení stejného poklesu hodnot HbA1c, navodil inzulin degludek významně nižší počet celkových [34] a zejména nočních hypoglykemií [34–36] u osob s diabetem 1. typu [35, 36] a 2. typu [34]. Právě nižší variabilita metabolického účinku může být značnou výhodou při titraci dávky nemocného a nabízí i vysvětlení pro rozdíl v četnosti výskytu hypoglykemických epizod pozorovaný v klinických studiích.
maximálního metabolického efektu GIRmax byla signifikantně nižší u degludeku (CV 18 %) ve srovnání s glarginem (CV 60 %), p < 0,0001, viz graf 5 [23]. I když je vždy potřebná značná opatrnost, pokud se data z experimentální situace přenášejí do reálné léčebné praxe, tak v tomto případě se předpokládané snížení rizika vzniku hypoglykemií při použití degludeku potvrdilo výsledky klinických studií. Opět ve srovnání s inzulinem glargin, při dosažení stejného poklesu hodnot HbA1c, navodil inzulin degludek významně nižší počet celkových [34] a zejména nočních hypoglykemií [34–36] u osob s diabetem 1. typu [35, 36] a 2. typu [34]. Právě nižší variabilita metabolického účinku může být značnou výhodou při titraci dávky nemocného a nabízí i vysvětlení pro rozdíl v četnosti výskytu hypoglykemických epizod pozorovaný v klinických studiích.
LY2605541 – využití hydrodynamické velikosti molekuly
Zcela odlišným mechanismem využitým pro tvorbu bazálního analoga inzulinu je modifikace molekuly připojením segmentu polyethylenglykolu (PEG). Touto změnou se výrazně změní její „hydrodynamická“ velikost, která je významným faktorem zpomalujícím vstřebávání z podkoží, a navíc i renální clearance. Molekula vyvíjeného bazálního analoga LY2605541 tedy obsahuje 20 kDA PEG jednotku připojenou k lysinu inzulinu lispro v pozici B28 ![Obr. 9 Design bazálního inzulinu LY2605541 – využití hydrodynamické velikosti molekuly; podle [37] – Hansen, et al., 2012.](https://www.remedia.cz/photo-a-29176---.jpg) prostřednictvím jeho ε-aminoskupiny. Pomocí dynamického rozptylu světla bylo zjištěno, že hydrodynamický průměr této molekuly je cca 7,8 ± 0,4 nm, což je 4násobek průměru molekuly inzulinu lispro a odpovídá tak průměru globulinu o molekulové hmotnosti 75 kDA, viz obr. 9 [37]. Tyto fyzikální parametry se promítly do farmakokinetiky tohoto inzulinového analoga vyjádřené výrazným posunem doby dosažení maximální plazmatické koncentrace (tmax, cca 25krát) a značně redukovanou clearance (10krát). Ve fázi I klinického zkoušení léčiva byly hodnoceny základní parametry pomocí euglykemických clampů, v nichž při použití různých dávek s.c. podaného léčiva byly průměry t1/2 v rozsahu 24,4–45,5 hod. a trvání účinku analoga na základě rychlosti infuze glukózy trvalo minimálně 36 hodin. Pokud byl LY2605541 podán intravenózně, byl jeho t1/2 zhruba 2,3 hodiny a bylo možné určit absolutní biologickou dostupnost při jeho s.c. podání (73 %). LY2605541 vykazoval nízkou míru intraindividuální variability (vyjádřenou CV %) < 18 % pro farmakokinetiku a < 32 % pro farmakodynamické parametry [38]. Další data o tomto inzulinovém analogu byla získána v rámci clampové studie za us
prostřednictvím jeho ε-aminoskupiny. Pomocí dynamického rozptylu světla bylo zjištěno, že hydrodynamický průměr této molekuly je cca 7,8 ± 0,4 nm, což je 4násobek průměru molekuly inzulinu lispro a odpovídá tak průměru globulinu o molekulové hmotnosti 75 kDA, viz obr. 9 [37]. Tyto fyzikální parametry se promítly do farmakokinetiky tohoto inzulinového analoga vyjádřené výrazným posunem doby dosažení maximální plazmatické koncentrace (tmax, cca 25krát) a značně redukovanou clearance (10krát). Ve fázi I klinického zkoušení léčiva byly hodnoceny základní parametry pomocí euglykemických clampů, v nichž při použití různých dávek s.c. podaného léčiva byly průměry t1/2 v rozsahu 24,4–45,5 hod. a trvání účinku analoga na základě rychlosti infuze glukózy trvalo minimálně 36 hodin. Pokud byl LY2605541 podán intravenózně, byl jeho t1/2 zhruba 2,3 hodiny a bylo možné určit absolutní biologickou dostupnost při jeho s.c. podání (73 %). LY2605541 vykazoval nízkou míru intraindividuální variability (vyjádřenou CV %) < 18 % pro farmakokinetiku a < 32 % pro farmakodynamické parametry [38]. Další data o tomto inzulinovém analogu byla získána v rámci clampové studie za us![Graf 6 Farmakokinetika a farmakodynamika LY2605541 u osob s diabetem 2. typu; podle [39] – Heise, et al., 2012. GIR – glucose infusion rate, rychlost infuze glukózy](https://www.remedia.cz/photo-a-29177---.jpg) táleného stavu (který odpovídá klinické praxi). Farmakokinetický ustálený stav byl navozen po 7–10 dnech podávání a poměr maximálních a údolních koncentrací byl < 1,5, což se projevilo téměř „bezvrcholovou“ rychlostí infuze glukózy v průběhu clampu a při těchto podmínkách se délka t1/2 pohybovala v rozmezí 44,7–75,5 hodiny (graf 6). Po dosažení ustáleného stavu bylo nutné u vyšetřovaných osob redukovat dávky prandiálního inzulinu a glykemie nalačno se posunula do pásma 3,33–5,5 mmol/l [39]. Výjimečnou vlastností LY2605541 je jeho hepatoselektivní působení, protože snáze proniká fenestracemi v jaterních sinusoidách než endotelem kapilár ve svalové či tukové tkáni. Právě relativní omezení účinku inzulinu v adipocytu spojené se snížením lipogeneze a naopak zvýšením míry lipolýzy se pravděpodobně promítá do pozorovaných změn hladin triglyceridů (↑), LDL cholesterolu (↑) a HDL cholesterolu (↓) a zvýšení hladin ALT a AST u léčených osob ve srovnání s inzulinem glargin (všechny ukazatele p < 0,02) [40]. Zároveň byl zaznamenán pokles hmotnosti léčených osob ve srovnání s glarginem [40, 41]. Nicméně celkový dopad těchto metabolických změn na kardiovaskulární ukazatele budou hodnotit probíhající studie fáze III klinického zkoušení.
táleného stavu (který odpovídá klinické praxi). Farmakokinetický ustálený stav byl navozen po 7–10 dnech podávání a poměr maximálních a údolních koncentrací byl < 1,5, což se projevilo téměř „bezvrcholovou“ rychlostí infuze glukózy v průběhu clampu a při těchto podmínkách se délka t1/2 pohybovala v rozmezí 44,7–75,5 hodiny (graf 6). Po dosažení ustáleného stavu bylo nutné u vyšetřovaných osob redukovat dávky prandiálního inzulinu a glykemie nalačno se posunula do pásma 3,33–5,5 mmol/l [39]. Výjimečnou vlastností LY2605541 je jeho hepatoselektivní působení, protože snáze proniká fenestracemi v jaterních sinusoidách než endotelem kapilár ve svalové či tukové tkáni. Právě relativní omezení účinku inzulinu v adipocytu spojené se snížením lipogeneze a naopak zvýšením míry lipolýzy se pravděpodobně promítá do pozorovaných změn hladin triglyceridů (↑), LDL cholesterolu (↑) a HDL cholesterolu (↓) a zvýšení hladin ALT a AST u léčených osob ve srovnání s inzulinem glargin (všechny ukazatele p < 0,02) [40]. Zároveň byl zaznamenán pokles hmotnosti léčených osob ve srovnání s glarginem [40, 41]. Nicméně celkový dopad těchto metabolických změn na kardiovaskulární ukazatele budou hodnotit probíhající studie fáze III klinického zkoušení.
Samotná „PEGylace“, tedy připojení molekuly polyethylenglykolu, je již ověřenou strategií, jak zvýšit terapeutické účinky proteinů [42]. Její použití v případě modifikace molekuly inzulinu navíc mění dostupnost přípravku v jednotlivých cílových tkáních. Teoretické snahy o změnu poměru účinku inzulinu v jaterní tkáni ve srovnání se svaly a tukovou tkání, o jejichž realizaci se již v minulosti neúspěšně usilovalo vývojem analoga thyroxyl-inzulinu a analoga tvořeného připojením dvou segmentů 9-fluorenylmethyloxykarbonylu k molekule inzulinu, tak nabývají praktické podoby [43].
Závěr
Vývoj inzulinových analog jistě nekončí, protože potřeby klinické praxe ve smyslu zjednodušení léčby (vyžadující vyrovnaný metabolický účinek po celých 24 hodin), snížení rizika hypoglykemií (umožňujícího snadné zahájení léčby a dobrou compliance), bezpečného použití u určitých skupin nemocných (např. s renálním selháním) a neutrálního vlivu na tělesnou hmotnost léčených osob ještě nejsou zcela naplněny. Inovace na poli farmakokinetiky inzulinů tak zůstanou nadále centrem zájmu výzkumu, a lze tedy navázat na odkaz Hagedorna, který mezi prvními ukázal, jak významně se odrazí změna farmakokinetických parametrů inzulinu (vývojem systému řízeného uvolňování) v jeho klinickém využití.
Seznam použité literatury
- [1] Joslin EP. The Treatment of Diabetes Mellitus, ed 4. Philadelphia, Lea & Febiger, 1928; pp 66–68, 80.
- [2] Hagedorn HC, Jensen BN, Krarup NB, et al. Protamine insulinate. JAMA 1936; 106: 177–180.
- [3] Krarup NB. Clinical Investigations Into the Action of Protamine Insulinate. Copenhagen, GEC Gad, 1935.
- [4] Felig P. Protamine Insulin Hagedorn´s Pioneering Contribution to Drug Delivery in the Management od Diabetes. JAMA 1984; 251: 393–396.
- [5] Scott DA, Fisher AM. The effect of zinc salts on the action of insulin. J Pharm Exp Ther 1935; 55: 206–216.
- [6] Krayenbuhl C, Rosenberg T. Crystalline protamine insulin. Rep Steno Memorial Hosp 1946; 1: 60–73.
- [7] Lindström T, Olsson PO, Arnquist HJ. The use of human ultralente is limited by great intraindividual variability in overnight plasma insulin profiles. Scand J Clin Lab Invest 2000; 60: 341–348.
- [8] Hildebrandt P, Berger A, Volund AA, et al. The subcutaneous absorption of human or bovine ultralente insulin formulations. Diabetic Med 1985; 2: 355–359.
- [9] Owens DR, Vora JP, Heding LG, et al. Human, porcine and bovine ultralente insulin: Subcutaneous administration in normal man. Diabetic Med 1986; 3: 316–319.
- [10] Adams MJ, Blundell TL, Dodson EJ, et al. Structure of rhombohedral 2 zinc insulin crystals. Nature 1969; 224: 491–495.
- [11] Zinman B, Tildsley H, Chasson JL, et al. Insulin lispro in CSII: results of a double-blind cross-over study. Diabetes 1997; 46: 440–443.
- [12] Markussen J. Engineering novel, prolonged acting insulins. In Hook JB, Pot G, eds. Protein design and the development of next therapeutics and vaccines. Plenum Publishing Co 1990; 19: 397–422.
- [13] Jørgensen S, Vaag A, Langkjaer L, et al. Novo/Sol basal: pharmacokinetics of a novel soluble long-acting soluble insulin analogue. BMJ 1989; 299: 415–419.
- [14] Scholtz HE, van Niekesk N, Mayer BH, et al. An assessment of the variability in the pharmacodynamics (glucose lowering effect) of HOE901 compared to NPH and ultralente human insulins using euglycaemic clamp technique. Diabetologia 1999; 42: A235.
- [15] Lepore M, Pampanelli S, Fanelli CG, et al. Pharmacokinetics of subcutaneous injection of long-acting human insulin analog glargine, NPH insulin, ultralente human insulin, and continuous subcutaneous infusio of insulin lispro. Diabetes 2000; 49: 2142–2148.
- [16] Heinemann L, Linkeschova R, Rave K, et al. Time-action profile of the long-acting insulin analog insulin glargine (HOE901) in comparison with those of NPH insulin and placebo. Diabetes Care 2000: 23: 644–649.
- [17] Yki-Jarvinen H, Dressler A, Ziemen M, et al. Less nocturnal hypoglycaemia and better post-dinner glucose control with bedtime insulin glargine compared with bedtime NPH insulin during insulin combination therapy in type 2 diabetes. HOE 901/3002 study group. Diabetes Care 2000; 23: 1130–1136.
- [18] Rosenstock J, Schwartz SL, Clark CM Jr, et al. Basal insulin therapy in type 2 diabetes: 28-week of insulin glargine (HOE 901) and NPH insulin. Diabetes Care 2001; 24: 631–636.
- [19] Owens DR, Coates PA, Luzio SD, et al. Pharmacokinetis of I-Labeled insulin glargine (HOE901) in healthy men-comparison with NPH insulin and the influence of different subcutaneous injection sites. Diabetes Care 2000; 23: 813–819.
- [20] Kurtzhals P. Engineering predictability and protraction in basal insulin analogue: the pharmacology of insulin detemir. Int J Obes 2004; 28 (Suppl. 2): S23–S28.
- [21] Markussen J, Havelund S, Kurtzhals P, et al. Soluble, fatty acid acylated insulins bind to albumin and show protracted actions in pigs. Diabetologia 1993; 39: 281–288.
- [22] Heise T, Nosek L, Ronn BB, et al. Lower within-subject variability of insulin detemir in comparison to NPH insulin and insulin glargine in people with type 1 diabetes. Diabetes 2004; 53: 1614–1620.
- [23] Heise T, Hermanski L, Nosek L, et al. Insulin degludes: four times lower pharmacodynamic variability than insulin glargine under steady-state conditions in type 1 diabetes. Diab Obes Metab 2012; 14: 859–864.
- [24] Jehle PM, Micheler C, Jehle DR, et al. Inadequate suspension of neutral protamine Hagedorn (NPH) insulin in pens. Lancet 1999; 354: 1604–1607.
- [25] Heise T, Pieber TR. Towards peakless, reproducible and long-acting insulins. An assessment of the basal analogues based on isoglycaemic clamp studies. Diab Obes Metab 2007; 9: 648–659.
- [26] Krüger P, Gilge G, Cabuk Y, et al. Cooperativity and intermediate states in the T R-structural transformation of insulin. Biol Chem Hoppe-Seyler. 1990; 371: 669–673.
- [27] Jonassen I, Havelund S, Ribel U, et al. Biochemical and physiological properties of a novel series of long-acting insulin analogs obtained by acylation with cholic acid derivatives. Pharm Res 2006; 23: 49–55.
- [28] Jonassen I, Havelund S, Hoeg-Jensen T, et al. Design of the Novel Protraction Mechanism of Insulin Degludec, an Ultra-long Acting Basal Insulin. Pharm Res 2012; 29: 2014–2114.
- [29] Kurtzhals P, Heise T, Strauss HM, et al. Multi-hexamer formation is the underlying mechanism behind the ultra-long glucose-lowering effect of insulin degludec. Diabetes 2011; 60 (Suppl 1): LB12.
- [30] Baker MP, Reynolds HM, Lumicisi B, et al. Immunogenicity of protein therapeutics: the key causes, consequences and challenges. Self Nonself 2010; 1: 301–322.
- [31] Heise T, Hövelmann U, Nosek L, et al. Insulin degludec has a two-fold longer half-life and a more consistent pharmacokinetic profile than insulin glargine. Diabetes 2011; 60 (Suppl 1): LB11.
- [32] Heise T, Nosek L, Bottcher SG, et al. Ultra-long-acting insulin degludec has a flat and stable glucose lowering effect in type 2 diabetes. Diab Obes Metab 2012; 14: 944–950.
- [33] Bott S, Tusek L, Jacobsen LV, et al. Insulin detemir under steady-state conditions: no accumulation and constant metabolic effect over time with twice daily administration in subjects with Type 1 diabetes. Diabet Med 2006; 23: 522–528.
- [34] Garber AJ, King AB, Del Prato S, et al. Insulin degludec, an ultra-longacting basal insulin, versus insulin glargine in basal-bolus treatment with mealtime insulin aspart in type 2 diabetes (BEGIN Basal-Bolus Type 2): a phase 3, randomised, open-label, treat-to-target non-inferiority trial. Lancet 2012; 379: 1498–1507.
- [35] Heller S, Buse J, Fisher M, et al. Insulin degludec, an ultra-longacting basal insulin, versus insulin glargine in basal-bolus treatment with mealtime insulin aspart in type 1 diabetes (BEGIN Basal-Bolus Type 1): a phase 3, randomised, open-label, treat-to-target non-inferiority trial. Lancet 2012; 379: 1489–1497.
- [36] Birkeland KI, Home PD, Wendisch U, et al. Insulin degludec in type 1 diabetes: a randomized controlled trial of a new-generation ultra-long-acting insulin compared with insulin glargine. Diabetes Care 2011; 34: 660–665.
- [37] Hansen RJ, Cutler JGB, Vick A, et al. LY26055441: Leveraging hydrodynamic size to develop a novel basal insulin. Scientific Sessions of the American Diabetes Association 2012; Poster 896-P.
- [38] Sinha VP, Howey DC, Xoon D, et al. Single-dose pharmacokinetics (PK) and glucodynamics (GD) of the novel long-acting basal insulin Ly2605541 in healthy subjects (Abstract). Scientific Sessions of the American Diabetes Association 2012; Poster 106-P.
- [40] Rosenstock J, Bergenstal RM, Blevins T, et al. Better Glycemic Control and Weight Loss With the Novel Long-Acting Basal Insulin LY2605541 Compared With Insulin Glargine in Type 1 Diabetes. Diabetes Care, Published online November 27, 2012.
- [41] Bergenstal RM, Rosenstock J, Akaraki RF, et al. A Randomized, Controlled Study of Once Daily LY2605541, a Novel Long-Acting Basal Insulin, Versus Insulin Glargine in Basal Insulin-Treated Patients with Type 2 Diabetes. Diabetes Care 2012; 35: 2140–2147.
- [42] Veronese FM, Pasut G. PEGylation, successful approach to drug delivery. Drug Discov Today 2005; 10: 1451–1458.
- [43] Shojaee-Moadie F, Eckey H, Jacson NC, et al. Novel hepatoselective insulin analogues: studies with covalently linked thyroxyl-insulin complexes. Diabet Med 1998; 15: 928–936.
- [44] Derewenda U, Derewenda Z, Dodson EJ, et al. Phenol stabilizes more helix in a new symmetrical zinc insulin hexamer. Nature 1989; 338: 594–596.
- [45] DeVries JH, Natrass M, Pieber TR. Refining basal insulin therapy: What have we learned in the age of analogues? Diabetes Metab Res Rev 2007; 23: 441–454.