Novinky v léčbě hemofilie
Souhrn:
Hemofilie je v současné době nemoc plně léčitelná s pomocí koncentrátů koagulačních faktorů VIII a IX vyráběných rekombinantní technologií nebo z lidské plazmy. Koncentráty dokáží zastavit krvácení a dají se použít i k profylaktické léčbě. Jejich společnou nevýhodou je však relativně krátký poločas účinku a aplikace výhradně intravenózní cestou. Větší či menší imunogenicita koncentrátů pak může vést ke vzniku inhibitoru (protilátky), který se může objevit až u 30 % těžkých hemofiliků a významně zkomplikovat jejich léčbu. Současný výzkum a vývoj léků na hemofilii se ubírá cestou prodloužení poločasu účinku koncentrátů koagulačních faktorů, odstranění nutnosti intravenózní aplikace a snížení imunogenicity. Ve vývoji jsou rovněž nové přípravky pro léčbu hemofiliků s inhibitorem, které by měly mít delší poločas účinku, vyšší účinnost a snazší laboratorní kontrolu. Souběžně s tím pokračuje intenzivní výzkum a příprava genové léčby, která již dospěla do stadia klinických studií s lidmi.
Key words:
hemophilia – replacement treatment – recombinant factor VIII and IX – half-life – prolonged half-life – bypassing agents – porcine recombinant factor VIII – gene therapy.
Summary:
Hemophilia is currently fully treatable by recombinant or plasma–derived factor VIII or IX concentrates. Coagulation factor concentrates are used to treat hemorrhages (on demand treatment) or for prophylactic treatment. Common disadvantages of both groups of factor concentrates are their short half life and purely intravenous administration. Stronger or weaker immunogenicity of clotting factor concentrates may lead to inhibitor development which occurs in up to 30% of severe hemophiliacs and significantly complicates their treatment. Current research focuses on development of prolonged half life clotting factor products, other than intravenous ways of their administration, and immunogenicity reduction. New concentrates for treatment of patients with inhibitors are also being developed which should have longer half life, better efficacy, and easier lab monitoring. At the same time, intensive research in gene therapy continues at level of clinical trials already.
Úvod
I když je hemofilie poměrně vzácná nemoc, hemofilie typu A se vyskytuje v poměru 1 : 10 000 porodů a hemofilie typu B v poměru 1 : 50 000 porodů, může se s ní i lékař „nehematolog“ ve své praxi setkat. Přitom je důležité, aby měl o tomto závažném onemocnění alespoň základní informace a mohl tak poskytnout nemocnému účinnou pomoc. Klasická 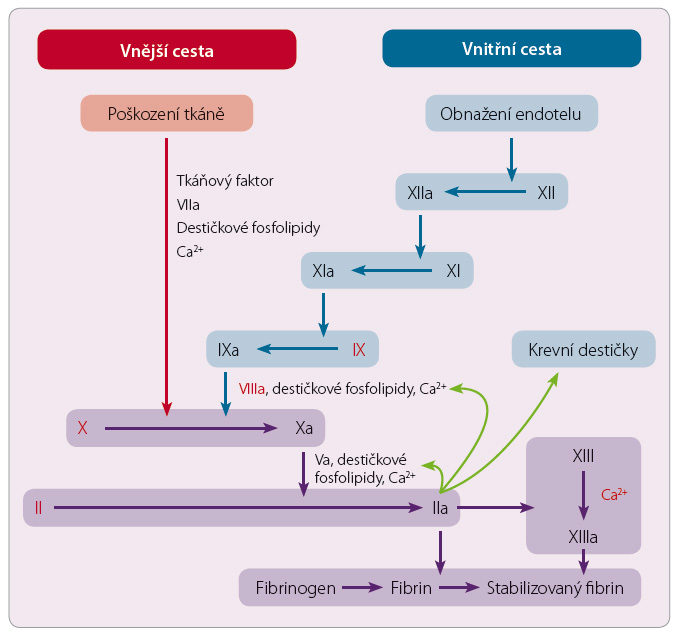 hemofilie A i hemofilie B se projevují excesivním krvácením a nelze je od sebe podle klinických projevů odlišit. Typickým místem krvácení jsou velké klouby a svaly, především dolních končetin, krvácení se pak projevuje bolestí, otokem a poruchou hybnosti v postiženém kloubu či svalu [1]. Prognóza hemofilie je díky novým diagnostickým a terapeutickým možnostem dnes natolik dobrá, že ji můžeme pokládat ve vyspělých zemích (včetně ČR) za plně kurabilní onemocnění [2]. Nemocní mají šanci dožít se takřka stejného věku jako jejich zdraví vrstevníci a k omezujícímu postižení pohybového aparátu dochází již zcela vzácně. Zásadní vliv na osud pacientů s hemofilií má v současnosti substituční léčba s dostatkem kvalitních koncentrátů koagulačních faktorů VIII a IX (FVIII a FIX), domácí a profylaktická substituční léčba a dobrá organizace péče o tyto pacienty na platformě Českého národního hemofilického programu (ČNHP). V současné době žije v ČR přibližně devět set hemofiliků, z toho čtvrtina dětí do 18 let.
hemofilie A i hemofilie B se projevují excesivním krvácením a nelze je od sebe podle klinických projevů odlišit. Typickým místem krvácení jsou velké klouby a svaly, především dolních končetin, krvácení se pak projevuje bolestí, otokem a poruchou hybnosti v postiženém kloubu či svalu [1]. Prognóza hemofilie je díky novým diagnostickým a terapeutickým možnostem dnes natolik dobrá, že ji můžeme pokládat ve vyspělých zemích (včetně ČR) za plně kurabilní onemocnění [2]. Nemocní mají šanci dožít se takřka stejného věku jako jejich zdraví vrstevníci a k omezujícímu postižení pohybového aparátu dochází již zcela vzácně. Zásadní vliv na osud pacientů s hemofilií má v současnosti substituční léčba s dostatkem kvalitních koncentrátů koagulačních faktorů VIII a IX (FVIII a FIX), domácí a profylaktická substituční léčba a dobrá organizace péče o tyto pacienty na platformě Českého národního hemofilického programu (ČNHP). V současné době žije v ČR přibližně devět set hemofiliků, z toho čtvrtina dětí do 18 let.
Schéma koagulační kaskády najdete pro připomenutí na obr. 1.
Substituční léčba
Do doby, než se stane klinicky použitelnou genová terapie, zůstanou hlavním léčebným prostředkem při léčení hemofilie substituční přípravky, ať již vyrobené z krevní plazmy, nebo rekombinantní technologií. Koncentráty koagulačních faktorů VIII a IX znamenaly v léčbě hemofilie doslova převrat a dramaticky změnily životy pacientů. S jejich pomocí lze provádět i nejsložitější chirurgické výkony, pacienti přestali být závislí na zdravotnických zařízeních a díky domácí a profylaktické léčbě mohou vést prakticky normální život. Zvláště u dětských pacientů znamená široká dostupnost substitučních koncentrátů obrovský pokrok. Substituční přípravky jsou vyráběny v lyofilizované formě a dají se skladovat při pokojové teplotě.
Léčba hemofilie se přesunula takřka plně do ambulance, hospitalizace pacienta je dnes nutná jen výjimečně, většinou pouze k chirurgickému výkonu. Pro běžná krvácení není nutné pacienty hospitalizovat a řadu krvácení zvládnou dokonce sami. Jak velký vliv má tento nový způsob léčby na psychiku a na sociální zařazení pacientů, není jistě třeba zdůrazňovat, nehledě na mnohem menší poškození tělesná. Nevýhodou substitučních přípravků je jejich značná cena, která se zvyšuje úměrně s jejich technologickou vyspělostí. Většina českých dětských hemofiliků je dnes léčena rekombinantními přípravky, zbytek vysoce čištěnými koncentráty vyráběnými z lidské plazmy. U dospělých je podíl pacientů léčených rekombinantními přípravky nižší. Čeští pacienti patří celosvětově mezi pouhou pětinu pacientů, kteří mají přístup k adekvátní substituci. Přípravků je dostatek, jsou plně hrazeny ze zdravotního pojištění a naše legislativní podmínky umožňují jejich použití jak k domácí, tak i k profylaktické léčbě.
Principy substituční léčby
U nekomplikovaných případů hemofilie typu A a i B zajistí hemostázu podání adekvátního množství FVIII nebo FIX obsaženého v substitučním přípravku. Celkové množství koncentrátu, které je nutno podat, závisí na několika okolnostech:
- na bazální hladině FVIII nebo FIX v pacientově plazmě,
- na závažnosti krvácení nebo chystaného chirurgického výkonu,
- na lokalizaci krvácení,
- na přítomnosti inhibitoru,
- na event. dalších poruchách hemostatického systému,
- na celkovém objemu pacientovy plazmy, potažmo tělesné hmotnosti,
- na koncentraci použitého substitučního přípravku,
- na biologickém poločase substitučního přípravku.
Všechny tyto faktory je třeba zvážit před výpočtem dávky substitučního přípravku. Při výpočtu dávky se vychází ze známého poznatku, že 1 IU FVIII podaná na 1 kg tělesné hmotnosti zvýší hladinu FVIII v pacientově plazmě o 1,5–2 %; 1 IU FIX podaná na 1 kg tělesné hmotnosti pak zvýší hladinu FIX v plazmě pacienta o 0,8–1 %. K dosažení účinné hemostázy je třeba, aby hladina FVIII dosáhla minimálně 30 %, v případě FIX minimálně 20 % [3]. U závažnějších krvácení je nutno dosáhnout hladin FVIII/FIX výrazně vyšších. Výše uvedené hodnoty jsou pouze orientační a pro správné dávkování substituční léčby je třeba určité klinické zkušenosti a zvážení všech okolností případu.
Substituční přípravky
Jak již bylo uvedeno výše, v současné době se k substituční léčbě hemofiliků používají v ekonomicky vyspělých zemích takřka výhradně koncentráty FVIII a FIX. Čerstvě zmražená plazma a kryoprecipitát přicházejí dnes v našich podmínkách v úvahu pouze jako vysloveně nouzové prostředky, v řadě chudých zemí však představují dosud jedinou možnost léčby. V České republice v současné době používáme u části pacientů koncentráty vysoké čistoty vyráběné z plazmy a je registrováno několik rekombinantních přípravků FVIII a FIX, které stojí na vrcholu pomyslné technologické pyramidy. Velká část českých hemofiliků typu A je léčena rekombinantními přípravky první až třetí generace, děti jsou léčeny rekombinantními přípravky pouze druhé a třetí generace.
Plazmatické koncentráty FVIII a FIX vysoké čistoty
Druhá generace plazmatických koncentrátů FVIII a FIX má specifickou aktivitu 50–200 IU FVIII na miligram proteinu, čehož je dosahováno čištěním na iontoměničích, chromatograficky nebo gelovou ultrafiltrací [6]. Tyto přípravky jsou proto označovány jako plazmatické koncentráty vysoké čistoty (high purity). Vysoká specifická aktivita umožnila koncentrovat stejné množství aktivity (vyjádřené v IU) do menšího objemu přípravku, než tomu bylo u přípravků střední čistoty. Většina přípravků je nyní k dispozici v objemu 2–5 ml, což usnadňuje (zvláště u dětí) intravenózní (i.v.) aplikaci.
Koncentráty FVIII vyráběné rekombinantní technologií
V současné době jsou používány tři genové technologie k výrobě těchto přípravků. První postup spočívá v produkci FVIII spolu s von Willebrandovým faktorem na tkáňových kulturách ovariálních buněk čínského křečka a v konečném produktu je von Willebrandův faktor odstraněn immunoafinní chromatografií. Při druhém postupu je získáván přímo čistý FVIII na tkáňových kulturách ledvinných buněk křeččích mláďat. Při třetím postupu jsou použity lidské embryonální ledvinné buňky. Biochemické, farmakokinetické a klinické vlastnosti takto vyrobených produktů jsou prakticky stejné jako u koncentrátů vyráběných z plazmy, specifická aktivita je však mnohonásobně vyšší a dosahuje více než 5 000 IU (u některých přípravků více než 10 000 IU) na miligram proteinu.
První generace rekombinantních přípravků obsahovala v médiu buněčné kultury zvířecí i lidské plazmatické proteiny, které byly přítomny i ve finálním produktu. Druhá generace rekombinantních přípravků obsahovala zvířecí nebo lidské proteiny v buněčném médiu, ne však již ve finálním produktu. Ani u těchto prvních dvou generací rekombinantních přípravků není tedy zcela eliminován kontakt s lidskou a zvířecí krví či tkání. Vedle lidských nákaz přenášených krví představují určité riziko také zatím neznámé zvířecí viry, které by mohly pocházet z buněčných kultur, a dále možnost senzibilizace příjemce koncentrátu myšími monoklonálními protilátkami užívanými k izolaci finálního produktu z buněčných kultur. Třetí generace rekombinantních přípravků FVIII již neobsahuje žádné plazmatické proteiny lidské ani zvířecí nejen ve finálním produktu, ale ani v buněčném médiu, produkt je stabilizován složitými glycidy. Tyto přípravky lze z mikrobiologického hlediska označit za první, stoprocentně bezpečný FVIII. Možnost přenosu lidské nebo zvířecí infekce je zde zcela vyloučena [4]. Současným vrcholem rekombinantní technologie je koagulační faktor VIII produkovaný lidskými buněčnými liniemi, který je již rovněž dostupný na farmaceutickém trhu.
Substituční léčba hemofilie B
Podobně jako v případě FVIII, i zde se vývoj ubíral cestou lyofilizovaných koncentrátů. Tyto koncentráty FIX jsou v lyofilizované podobě dlouhodobě stabilní i při pokojové teplotě, rozpouštějí se v malém množství rozpouštědla, takže při jejich použití nehrozí objemové přetížení pacienta. Jejich velkou nevýhodou je však poměrně značná trombogennost při vyšším dávkování, která je způsobena přítomností malých množství aktivovaných koagulačních faktorů, zejména FX. Posledně jmenovanou nevýhodu odstraňují moderní vysoce čištěné koncentráty FIX, které neobsahují příměs aktivovaných koagulačních faktorů a jejich trombogennost je velmi nízká. U dětí trpících hemofilií typu B jsou v České republice v současné době používány výhradně vysoce čištěné plazmatické koncentráty FIX. V zahraničí je na trhu a v běžném klinickém použití rovněž první rekombinantní koncentrát FIX třetí generace [5].
Bezpečnost substitučních přípravků
Bezpečnost substitučních přípravků pro léčbu hemofilie se dnes posuzuje ze dvou hledisek. Jedním, a podle mého názoru tím důležitějším, je mikrobiologická bezpečnost, druhým pak imunogenicita neboli schopnost indukovat tvorbu protilátky – inhibitoru. Z hlediska mikrobiologické bezpečnosti představují vrchol nabídky rekombinantní přípravky [7], z hlediska schopnosti indukovat tvorbu inhibitoru probíhá široká diskuse, zda rekombinantní přípravky nejsou více imunogenní než přípravky vyráběné z plazmy [8,9].
Domácí a profylaktická léčba hemofilie
V naší republice je domácí léčba legislativně upravena Věstníkem MZ ČR č. 3/1992. Pacienti dostávají substituční přípravky v hemofilických centrech, na odděleních klinické hematologie či na transfuzních odděleních a v případě potřeby si je aplikují sami, s pomocí rodiny nebo jim je v některých případech aplikuje praktický lékař pro děti a dorost. Podle údajů celostátního registru pacientů s hemofilií ČNHP využívá výhod domácí léčby 60 % všech dětských hemofiliků; u pacientů s těžkou formou nemoci je to takřka 100 %. S možností domácí léčby úzce souvisí problematika profylaktické léčby hemofilie. Jejím hlavním úkolem je zabránit vzniku invalidizující hemofilické artropatie u těžkých forem hemofilie [10]. K trvalému udržení žádoucích hodnot, tedy převyšujících 1 %, je nutné podávat konvenční FVIII obvykle 2–3× týdně a FIX 2× týdně. V poslední době se názory na podávání profylaktické substituce mění a do popředí se dostává individualizovaný (tzv. tailored) způsob profylaxe [11].
Nové možnosti léčby
Zásadní nevýhodou všech dosud užívaných substitučních přípravků je jejich relativně krátký poločas účinku. Poločas účinku FVIII se pohybuje mezi 8–12 hodinami, poločas účinku FIX mezi 18–24 hodinami. Abychom při profylaktické léčbě udrželi hladinu FVIII trvale vyšší než 1 %, musíme koncentrát FVIII podávat minimálně 3× týdně a v některých případech ob den. Koncentrát FIX je nutno podávat minimálně 2× týdně. Netřeba jistě zdůrazňovat, co to znamená zvláště pro dětské pacienty. Značná pozornost je tedy věnována vývoji přípravků s prodlouženou dobou účinku.
Další sledovanou vlastností užívaných přípravků je jejich imunogenicita a schopnost vyvolat tvorbu inhibitoru (protilátky proti zevně dodávanému FVIII/IX). Snahou výrobců substitučních přípravků v současné době je proto vyvinout koncentráty s prodlouženou dobou účinku a s nižší imunogenicitou, než je tomu u současných přípravků. Samostatnou kapitolou je potom genová léčba hemofilie.
Koncentráty FVIII/FIX s prodlouženou dobou účinku
Schopnost koncentrátů působit v hemostáze delší dobu vede k prodloužení intervalu mezi jednotlivými infuzemi při ![Obr. 2 Technologie používané k prodloužení biologického poločasu FVIII a FIX; podle [32] – Mannucci, et al., 2014. Fc – fragment crystallizable, konstantní část, která neváže antigen; PEG – polyethylenglykol * Modifi kace molekuly koagulačního faktoru probíhá na úrovni DNA, jsou fúzovány geny koagulačního faktoru a Fc, nikoli bílkoviny. ** Molekuly PEG jsou z organismu eliminovány přirozenou cestou (moč, stolice), nekumulují se v organismu.](https://www.remedia.cz/photo-a-30598---.jpg) profylaktické léčbě a k omezení počtu nutných i.v. injekcí. Řada takových přípravků se v současné době nachází ve stadiu klinického zkoušení a první z nich je již i v prodeji. Prodloužení účinku lze dosáhnout několika způsoby (obr. 2).
profylaktické léčbě a k omezení počtu nutných i.v. injekcí. Řada takových přípravků se v současné době nachází ve stadiu klinického zkoušení a první z nich je již i v prodeji. Prodloužení účinku lze dosáhnout několika způsoby (obr. 2).
Fúze s Fc doménou
Fúze biologicky aktivního proteinu s Fc doménou lidského IgG1 je spojena s ochranou proteinu před lyzosomální degradací a s prodloužením jeho biologického účinku. Rekombinantně vyrobený faktor VIII zbavený B domény (B‑domain‑deleted /BDD/) je kovalentní vazbou navázán na Fc doménu lidského IgG1. Ve studiích s těmito přípravky je prokazováno 1,6–1,7násobné prodloužení času eliminace a biologického poločasu FVIII‑Fc až na 19 hodin [12]. Podobně ošetřená přirozená molekula FIX vykazuje až trojnásobné prodloužení doby účinku a prodloužení biologického poločasu až na 82 hodin [13].
Fúze s albuminem
Albumin je v krvi přirozeně se vyskytující protein a jeho biologický poločas zde dosahuje více než 20 dnů. Je široce používán jako bílkovinný stabilizátor při výrobě řady léčiv. Rekombinantní FIX s navázaným rekombinantním albuminem (rFIX‑FP) dosahuje poločasu až 102 hodin, což je 4,3krát více než u běžných přípravků FIX [14]. Přípravek tohoto typu zkoušíme na naší klinice u jednoho chlapce s těžkou hemofilií typu B a k účinné profylaktické léčbě stačí aplikace 40 IU/kg podaných jednou za sedm dní. První rekombinantní přípravek FVIII a FIX tohoto typu je již v prodeji.
Pegylace
Pegylace znamená kovalentní připojení polyethylenglykolu (PEG) k proteinu, k peptidu nebo k malé lékové molekule. Pegylace prodlužuje biologický poločas bílkovin několika mechanismy: a) zvětšením jejich molekuly, b) snížením jejich citlivosti k proteolytickému štěpení a k degradaci a c) změnou vlastností povrchového elektrického náboje dochází k interferenci s procesy clearance. Pegylací ošetřený FVIII má ve srovnání s běžnými přípravky přibližně dvojnásobně prodloužený poločas účinku [15]. Probíhající klinické studie s pegylovanými přípravky zkoumají rovněž potenciální vedlejší účinky pegylace [16]. První pegylovaný rekombinantní přípravek FVIII je již na trhu ve Spojených státech amerických.
Polysialyzace
Vazba koagulačního proteinu na kyselinu polysialovou vede k podobnému efektu jako pegylace a prodlužuje poločas účinku FVIII/FIX [17,18].
Inhibice TFPI
Inhibitor TFPI (tissue factor pathway inhibitor) v plazmě moduluje koagulaci spuštěnou tkáňovým faktorem (TF); TFPI váže a inhibuje komplexy TF‑FVII/FVIIa. Pokud je TFPI inhibován, dochází ke zvýšené generaci fibrinu. K inhibitorům TFPI patří aptamery (jednovláknové nukleové kyseliny) a fucoidany (polysacharidy podobné heparinům bez antikoagulačních vlastností) extrahované z mořských řas. Aptamery lze podávat podkožně, fucoidany podkožně, a dokonce i perorálně. Jde o nadějné látky, které by umožnily léčbu hemofilie bez nutnosti i.v. injekcí [19–21].
Koncentráty FVIII/FIX se sníženou imunogenicitou
Vývoj substitučních přípravků se sníženou imunogenicitou je zaměřen na snížení počtu pacientů s inhibitorem (protilátkou) FVIII/FIX. V současné době se jedná o nejzávažnější komplikaci hemofilie, která postihuje asi 30 % pacientů s těžkou hemofilií A a 3 % pacientů s těžkou hemofilií B. Přítomnost inhibitoru znemožňuje léčbu běžnými koncentráty, ty jsou protilátkou neutralizovány, a léčba je proto méně účinná a extrémně nákladná.
Rekombinantní FVIII s odstraněnou B doménou (BDD) produkovaný lidskou buněčnou linií
Jedná se o rekombinantní faktor VIII produkovaný buněčnými liniemi lidských embryonálních ledvinných buněk. Takto vyrobený přípravek, který je již v prodeji a v klinickém používání, vykazuje stejné vlastnosti jako plazmatické přípravky, normální vazbu von Willebrandova faktoru a fosfolipidů, aktivaci a tvorbu fibrinu a FXa. Afinita k von Willebrandovu faktoru je zhruba o 40 % vyšší ve srovnání s ostatními rekombinantními přípravky FVIII, což vede k jeho větší stabilitě, k prodlouženému poločasu účinku a ke snížené imunogenicitě [22,23].
Jednořetězcový rekombinantní FVIII
Rekombinantní faktor VIII se skládá ze dvou spojených proteinových řetězců, lehkého a těžkého, které lze různými způsoby oddělit. Lze však připravit jednořetězcový rekombinantní FVIII, kde jsou spojeny lehký a těžký řetězec kovalentní vazbou. Takto připravený jednořetězcový faktor vykazuje zvýšenou molekulární integritu i stabilitu a má zvýšenou afinitu k von Willebrandovu faktoru, což může vést k jeho snížené imunogenicitě.
Léčivé přípravky používané v terapii pacientů s inhibitorem proti FVIII/FIX
V léčbě pacientů s inhibitorem se v současnosti používají tzv. bypassing přípravky, které aktivují FX na FXa i bez přítomnosti FVIII/FIX. V klinickém použití jsou v nezměněné podobě již řadu let přípravky aktivovaného protrombinového komplexu (activated prothrombin complex concentrate, aPCC) a rekombinantní aktivovaný FVII (rFVIIa). I v této oblasti se ale vývoj nezastavil a jsou vyvíjeny nové molekuly rFVIIa, které by měly mít lepší farmakologické vlastnosti, zvláště prodloužení účinku, ten je u stávajícího přípravku velmi krátký. K prodloužení účinku a k výraznější tvorbě trombinu vede vazba s pegylovanými liposomy, k rychlejšímu a déle trvajícímu účinku vede analog rFVIIa vatreptacog alfa [24]. Zkoumány jsou rovněž varianty rFVIIa vzniklé různými arteficiálními změnami aminokyselin v řetězci. V klinickém zkoušení je rovněž transgenní lidský rFVIIa získávaný z mléka králíků, který by měl být méně imunogenní, dostupnější a levnější než současný přípravek.
Bispecifická protilátka
V normální hemostáze působí FVIII jako kofaktor a umožňuje aktivaci faktoru X (FXa) aktivovaným FIX. Pokud je FVIII nedostatek nebo je neúčinný (neutralizace inhibitorem), aktivace FX nemůže proběhnout. Nahradíme‑li ale FVIII bispecifickou protilátkou namířenou proti FIXa na jedné straně a proti FX na straně druhé, dojde k prostorovému sblížení obou faktorů a k následné aktivaci FX na FXa. Bispecifická protilátka zde nahrazuje funkci FVIII jako kofaktoru [25]. Tato látka má biologický poločas 4–5 týdnů a aplikuje se pokusným zvířatům (opice) a nyní již i lidem [26] podkožně. Uspokojivě funguje jak u inhibitoru, tak i u prosté hemofilie A.
Rekombinantní vepřový FVIII
Vepřový faktor VIII je staronovým prostředkem pro léčbu hemofiliků s vysokým titrem inhibitoru a pro léčbu pacientů se získanou hemofilií. Plazmatický vepřový FVIII byl s úspěchem používán již před několika desítkami let. Využívalo se nízké zkřížené reaktivity s protilátkami proti lidskému inhibitoru a klinické odpovědi, která byla při léčbě tímto přípravkem lepší než při léčbě tzv. bypassing přípravky. Výhodou byla rovněž možnost stanovení hodnoty porcinního FVIII a monitorace léčby, zatímco u tzv. bypassing přípravků je laboratorní sledování léčby velmi obtížné. V roce 2004 byl však přípravek stažen z trhu pro přenos prasečího parvoviru. Vývoj rekombinantního vepřového FVIII tento problém vyřešil a přípravek je již k dispozici na americkém trhu [27].
Genová léčba hemofilie
Naději pro všechny pacienty trpící hemofilií představuje do budoucna genová léčba. Odhalení struktury a klonování genu pro FVIII a FIX na počátku osmdesátých let minulého století umožnilo nejenom výrobu rekombinantních substitučních přípravků, ale vytvořilo i základní teoretický předpoklad pro tuto léčbu. Hemofilie s její jednoduchou dědičností a snadným sledováním účinku léčby (hodnota FVIII/FIX) je ideálním modelem pro genovou terapii. Po počátečním entusiasmu se ukázalo, že genová léčba má řadu úskalí a její zavedení nebude tak rychlé, jak se předpokládalo. Po celé řadě experimentálních prací na zvířatech byly již v roce 1999 zahájeny v USA, a později v dalších zemích, první klinické zkoušky u lidí [28]. Jako schůdnější se ukázala genová léčba hemofilie B a v současné době lze shrnout, že dospělý nemocný s těžkou hemofilií typu B, s negativitou HCV RNA (ribonukleová kyselina viru hepatitidy C) a bez protilátek proti AAV (adeno‑associated virus) [29], vektoru přenosu, může podstoupit intravenózní infuzi vektoru AAV [30] s genem přirozeného FIX. Po infuzi lze očekávat nejméně tři roky trvající sekreci FIX s dosažením hladiny kolem 5 %.
Jedná se o výrazný úspěch, kdy se těžká hemofilie mění v lehkou a dramaticky se tak mění klinický průběh nemoci. V současnosti probíhá několik klinických studií u hemofilie B, kde lze očekávat podobné výsledky. Genová léčba hemofilie A je obtížnější, což je dáno především větší velikostí genu pro FVIII a jeho obtížnější inkorporací do hostitelské buňky [31]. Pokusy na zvířatech však dokazují zvýšení sekrece FVIII po léčbě na 3–8 %. Genová léčba postihuje somatické buňky a může zlepšit zdravotní stav, nebo i vyléčit daného jedince. Protože ale nepostihuje zárodečné buňky, nezabrání přenosu defektního genu na pacientovy dcery, které se tak, stejně jako je tomu dosud, stanou přenašečkami. I když klinické studie přinášejí zajímavé výsledky, je třeba k zavedení genové léčby do praxe vyřešit ještě celou řadu problémů a nejedná se o metodu použitelnou v blízké budoucnosti. O skutečném vyléčení hemofilie pak budeme moci hovořit teprve tehdy, až bude k dispozici genová léčba postihující i zárodečné buňky.
Závěr
Jak je z uvedeného přehledu patrno, výzkum se soustředí především na prodloužení účinku FVIII a IX a na nalezení takových přípravků, které by nevyžadovaly intravenózní aplikaci. Oba směry mohou vést k uvedení nových přípravků, které výrazně zjednoduší a zpříjemní léčbu hemofilie, do širší praxe. Ve hře bude jistě dostupnost nových přípravků, především z ekonomického hlediska. Drahý vývoj se musí farmaceutickým firmám zaplatit, a tak cena nebude jistě nízká. K současné léčbě má přístup pouze asi pětina hemofiliků v bohatých zemích světa (mezi něž patří i ČR), takže lze předpokládat, že nové léky na hemofilii ještě více rozevřou pomyslné nůžky mezi bohatým severem a chudým jihem.
Práce byla vytvořena s podporou VZ FNM č. MZO 00064203.
Seznam použité literatury
- [1] World Federation of Hemophilia: Guidelines for the management of Hemophilia. World Federation of Hemophilia, Montréal, Québec 2005.
- [2] Komrska V. Hemofilie v dětském věku, in: Jan Starý et al., Dětská hematologie. Praha: Galén, 2005; s. 141–142.
- [3] Blatný J, Komrska V, Matýšková M, et al. Hemofilie – doporučené postupy při léčbě krvácivých epizod, in: Doporučené postupy, ČHS ČLS JEP, Český spolek pro trombózu a hemostázu, Pracovní skupina pro standardy ČNHP, květen 2009 (účelová publikace).
- [4] Pipe S. Management of Hemophilia in the Midst of Emerging Pathogens: A Societal Perspective. Seminars in Hematology 2006; 43 (Supplement 3): S1–S3.
- [5] Berntorp E, Shapiro AD, Waters J. The international factor IX treatment network survey (Letter to the editor). Haemophilia 2012; 18: e60–e62.
- [6] Medical and Scientific Advisory Council (MASAC) of the National Hemophilia Foundation (USA) – Document #177, New York, October 14 2006.
- [7] Valentino LA, Oza VM. Blood Safety and the Choice of Anti Hemophilic Factor Concentrate. Pediatr Blood Cancer 2006; 47: 245–254.
- [8] Gouw SC, Van der Bom JG, Auerswald G, et al. Recombinant versus plasma derived factor VIII products and the development of inhibitors in previously untreated patients with severe hemophilia A: the CANAL cohort study. Blood 2007; 109: 4693–4697.
- [9] Goudemand J, Rothschild C, Demiguel V, et al. Influence of the type of factor VIII concentrate on the incidence of factor VIII inhibitors in previously untreated patients with severe hemophilia A. Blood 2006; 107: 46–51.
- [10] Gringeri A, Lundin B, Von Mackensen S, et al. A randomized clinical trial of prophylaxis in children with hemophilia A (the ESPRIT Study). J Thromb Haemost 2011; 9: 700–710.
- [11] Kurnik K, Bidlingmaier C, Engl W, et al. New early prophylaxis regimen that avoids immunological danger signals can reduce FVIII inhibitor development. Haemophilia 2010; 17: 256–262.
- [12] Mahlangu J, Powell JS, Ragni MV, et al. Phase 3 study of recombinant factor VIII Fc fusion protein in severe hemophilia A. Blood 2014; 123: 317–325.
- [13] Powell JS, Pasi KJ, Ragni MV, et al. Phase 3 study of recombinant factor IX Fc fusion protein in hemophilia B. N Engl J Med 2013; 369: 2313–2323.
- [14] Santagostino E, Martinowitz U, Lissitchkov T, et al. Long acting recombinant coagulation factor IX albumin fusion protein (rIX FP) in hemophilia B: results of a phase 3 trial. Blood 2016; pii: blood 2015 09 669234.
- [15] Ivens IA, Baumann A, McDonald TA, et al. PEGylated therapeutic proteins for hemophilia treatment: a review for hemophilia caregivers. Haemophilia 2013; 19: 11–20.
- [16] Stidl R, Fuchs S, Bossard M, et al. Safety of PEGylated recombinant human full length coagulation factor VIII (BAX 855) in the overall context of PEG and PEG conjugates. Haemophilia 2016; 22: 54–64.
- [17] Pipe SW. Hemophilia: new protein therapeutics. Hematol Am Soc Hematol Educ Program 2010; 116: 270–279.
- [18] Wu J, Lu S, Zheng Z, et al. Modification with polysialic acid PEG copolymer as a new method for improving the therapeutic efficacy of proteins. Prep Biochem Biotechnol 2016, Feb 1.
- [19] Prasad S, Lillicrap D, Labelle A, et al. Efficacy and safety of a new class hemostatic drug candidate, AV513, in dogs with hemophilia A. Blood 2008; 111: 672–679.
- [20] Liu T, Scallan CD, Broze GJ Jr, et al. Improved coagulation in bleeding disorders by Non Anticoagulant Sulfated Polysacharides (NASP). Thromb Haemost 2006; 95: 68–76.
- [21] Shetty S, Ghosh K. Novel therapeutic approaches for haemophilia. Haemophilia 2015; 21: 152–161.
- [22] Lissitchkov T, Hampton K, von Depka M, et al. Novel, human cell line derived recombinant factor VIII (human cl rhFVIII, Nuwiq) in adults with severe haemophilia A: efficacy and safety. Haemophilia 2015 Aug 28, doi: 10.1111/hae.12793.
- [23] Klukowska A, Szczepanski T, Vdovin V, et al. Novel, human cell line derived recombinant factor VIII (Human cl rhFVIII, Nuwiq) in children with severe haemophilia A: efficacy, safety and pharmacokinetics. Haemophilia 2015 Sep 14, doi: 10.1111/hae.12797.
- [24] De Paula EV, Kavakli K, Mahlangu J, et al. Recombinant factor VIIa analog (vatreptacog alfa/activated/) for treatment of join t bleeds in hemophilia patients with inhibitors: a randomised controlled trial. J Thromb Haemost 2012; 10: 81–90.
- [25] Kitazawa T, Igawa T, Sampei Z, et al. A bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nat Med 2012; 18: 1570–1574.
- [26] Uchida N, Sambe T, Yoneyama K, et al. A first in human phase 1 study of ACE910, a novel factor VIII mimetic bispecific antibody, in healthy subject. Blood 2015 Dec 1, pii: blood 2015 06 650226.
- [27] Lillicrap D, Schiviz A, Apostol C, et al. Porcine recombinant factor VIII (Obizur, OBI 1, BAX801): product characteristics and preclinical profile. Haemophilia 2015 Aug 17, doi: 10.1111/hae.12784.
- [28] George LA, Fogarty PF. Gene therapy for hemophilia: past, present and future. Semin Hematol 2016; 53: 46–54.
- [29] Van der Loo JC, Wright JF. Progress and challenges in viral vector manufacturing. Hum Mol Genet 2015 Oct 30, doi: 10.1093/hmg/ddv451.
- [30] High KA, Anguela XM. Adeno associated viral vectors for the treatment of haemophilia. Hum Mol Genet 2015 Nov 27, pii: ddv475.
- [31] Sabatino DE, Lange AM, Altynova ES, et al. Efficacy and safety of long term prophylaxis in severe hemophilia hemophilia A dogs following liver gene therapy using AAV vectors. Mol Ther 2011; 19: 442–449.
- [32] Mannucci PM, Franchini M. Expert Opin Emerg Drugs 2014; 19: 407–414.