Postavení ruxolitinibu v léčbě myeloproliferativních chorob po sedmi letech od jeho schválení do klinické praxe
Souhrn:
Bělohlávková P. Postavení ruxolitinibu v léčbě myeloproliferativních chorob po sedmi letech od jeho schválení do klinické praxe. Remedia 2019; 29: 95–98.
Nejčastěji se vyskytující Ph negativní (Philadelphia chromosome‑negative) myeloproliferativní choroby (MPN) představují polycythaemia vera (PV), esenciální trombocytemie (ET) a primární myelofibróza (PMF). Objevy somatických mutací JAK2V617F, CALR a MPL se staly důležitými prvky pro pochopení patogeneze PMF a tyto mutace jsou odpovědné za vlastní fenotyp onemocnění. Ze skupiny inhibitorů JAK1/2 byl v roce 2011 prvním zástupcem ruxolitinib, který byl schválen americkým Úřadem pro kontrolu potravin a léčiv (FDA) pro léčbu PMF. Hematologická toxicita a zvýšené riziko infekcí jsou nejčastějšími nežádoucími účinky této léčby. I když se uskutečnily studie s dalšími inhibitory JAK1/2 (fedratinib, momelotinib, pacritinib), žádný další inhibitor do terapie MPN není dosud schválen.
Summary:
Belohlavkova P. Ruxolitinib in the treatment of myeloproliferative diseases after seven years since the approval for clinical practice. Remedia 2019; 29: 95–98.
The most common Philadelphia‑negative (Ph‑) myeloproliferative diseases (MPD) are polycythemia vera (PV), essential thrombocythemia (ET) and primary myelofibrosis (PMF). The discoveries of JAK2V617F, CALR, and MPL somatic mutations have become important elements for understanding the pathogenesis of PMF, and these mutations are responsible for their own phenotype of the disease. In the JAK1/2 inhibitor group, ruxolitinib was the first drug to be approved by the Food and Drug Administration (FDA) for the treatment of PMF in 2011. The hematologic toxicity and increased risk of infections are the most common side effects of the treatment with ruxolitinib. Although studies with other JAK1/2 inhibitors (fedratinib, momelotinib, pacritinib) have been performed, no other ihibitors are approved for the therapy of MPD yet.
Key words: JAK1/2 inhibitors, polycythemia vera, primary myelofibrosis,ruxolitinib
Úvod
Ph negativní myeloproliferativní choroby (Ph MPN) zahrnují několik klinických podjednotek, ale nejčastější nemoci představují polycythaemia vera (PV), esenciální trombocytemie (ET) a primární myelofibróza (PMF). Příčinou rozvoje těchto onemocnění je klonální poškození pluripotentní kmenové buňky, kdy dochází k proliferaci jedné nebo více myeloidních linií, ale je zachována jejich schopnost vyzrávat. Dalším společným rysem těchto onemocnění je zvýšená aktivace signální dráhy JAK STAT, která dává příslušným progenitorovým kmenovým buňkám impulz k proliferaci. Genetickým podkladem, který se spolupodílí na složité etiopatogenezi chorob, je přítomnost mutace genů pro tyrozinovou kinázu JAK2, kalretikulin (CALR) či trombopoetinový receptor (MPL) [1,2]. Jedná se o tzv. somatické mutace, které jsou zodpovědné za vlastní fenotyp chorob. Přítomnost těchto mutací je také využívána k diagnostice chorob (především u PV) a současně nese svůj klinický význam. Například prognóza pacientů „triple negativních“ (JAK2, CALR a MPL nemutovaných) u PMF je nepříznivá [3].
Incidence klasických Ph MPN se pohybuje u PV v rozmezí 0,2−2,2, u ET mezi 0,1−2,6 a u PMF mezi 0,3−1,0 na 100 000 obyvatel a rok. Medián věku pacientů v době diagnózy PV, ET a PMF je udáván kolem 60, 60 a 65 let. Onemocnění PV a PMF se vyskytují o něco častěji u mužů, ET je naopak poněkud častější u žen [4]. Přežívání pacientů s PV je ve srovnání s běžnou populací zkráceno a pohybuje se mezi 14−24 lety v závislosti na rizikových faktorech u daného jedince. Naopak celkové přežití u pacientů s ET by mělo být srovnatelné s ostatní populací [5,6].
Pacienti s PMF mají velmi variabilní prognózu (2,5–10 let) závisející na přítomnosti rizikových faktorů. V současné době při diagnóze PMF užíváme mezinárodní prognostický skórovací systém (IPSS) z roku 2009, ve kterém je hodnoceno pět rizikových faktorů (věk, anémie, počet leukocytů a blastů v periferii, přítomnost celkových příznaků) [7]. Podle výpočtu můžeme pacienty rozdělit do čtyř skupin: nízké riziko, střední 1, střední 2 a vysoké riziko s mediány přežití 135, 95, 48 a 27 měsíců. V roce 2010 byl publikován dynamický mezinárodní prognostický skórovací systém (DIPSS), jehož výhodou je možnost použití i v průběhu choroby. Ve snaze více zpřesnit prognózu pacientů byl tento prognostický systém rozšířen o tři rizikové faktory (transfuzní závislost, nepříznivý karyotyp a počet trombocytů) a je označován jako DIPSS plus [8,9].
Cílem léčby PV a ET je normalizace parametrů v krevním obraze a snížení rizika vzniku trombotických a krvácivých komplikací. Základní léčebné metody u PV představuje venepunkce, cytoreduktivní léčba (hydroxyurea, ev. anagrelid u trombocytemie) či léčba interferonem. Nejsou li kontraindikace, měla by být zahájena současně antiagregační léčba kyselinou acetylsalicylovou. Terapeutický přístup k ET zahrnuje opět antiagregační a cytoreduktivní léčbu (hydroxyurea, anagrelid) podle zhodnocení rizikovosti pacienta [10,11].
Pacienti s PMF mladší 65 (70) let s rizikem dle IPSS/DIPSS středním 2 a vysokým, kteří nemají závažné komorbidity, by měli být indikováni k alogenní transplantaci krvetvorných buněk (haematopoietic stem cell transplantation, HSCT). U ostatních pacientů se snažíme zmírnit celkové příznaky a ovlivnit dominující obtíže, které mohou vyplývat z anémie, splenomegalie. Léčba danazolem, erytropoetinem či kombinace nízkých dávek thalidomidu s nižší dávkou kortikoidu může být účinná u anémie. Cytolytická léčba (hydroxyurea) je indikována především u hyperproliferativních forem choroby [10,11].
K ovlivnění splenomegalie můžeme použít hydroxyureu, avšak pozitivní vliv této léčby je pozorován asi u třetiny pacientů a bývá často přechodný nebo je doprovázen nežádoucími účinky. Dříve se častěji prováděla i splenektomie, avšak tento výkon je provázen vysokým rizikem perioperačních komplikací včetně mortality a pětileté přežití pacientů po splenektomii se pohybuje kolem 20 %. Další možností ovlivnění splenomegalie je ozáření sleziny, avšak léčebný efekt je opět pouze přechodný s nebezpečím rozvoje protrahované pancytopenie, která je popisována až u 44 % nemocných (u 26 % velmi těžká forma) [12,13].
Ruxolitinib
Ruxolitinib (RUXO) je chemicky (R) 3 (4 (7H pyrrolo[2,3 d]pyrimidin 4 yl) 1H pyrazol 1 yl) 3 cyclopentylpropannitril fosfát s molekulovou hmotností 404,36 kDa. Je rozpustný ve vodných roztocích při pH 1−8. Absorpce RUXO je velmi rychlá, maximální sérové koncentrace je dosaženo již za 2 hodiny. Udávaný biologický poločas pro 100mg tablety je 3 hodiny a z tohoto důvodu je preferováno dávkování 2× denně. Ruxolitinib je metabolizován pomocí enzymů CYP3A4, o něco méně pomocí CYP2C9. Je tedy nutné počítat s možnými interakcemi, kdy např. flukonazol výrazně zvyšuje koncentraci RUXO v plazmě.
Ruxolitinib je tyrozinkinázový inhibitor JAK1/2, který svým účinkem zabraňuje aktivaci signálních drah JAK STAT. Těmito mechanismy dochází ke sníženému uvolňování celé řady prozánětlivých cytokinů a ke snížení proliferativní aktivity [14].
Ruxolitinib u primární
myelofibrózy
První klinická studie fáze I/II (NCT00509899) u pacientů s PMF prokázala jako maximálně tolerovanou dávku 25 mg RUXO podávaných dvakrát denně s velmi slibným efektem na ovlivnění splenomegalie a celkových příznaků. Následovaly dvě studie fáze III COMFORT I a COMFORT II, do kterých bylo zařazeno 155 a 146 pacientů s rizikem dle IPSS charakterizovaným jako střední 2 a vysoké. Tyto studie porovnávaly RUXO s placebem nebo s nejlepší možnou dostupnou léčbou (BAT, best available therapy). Ve studii COMFORT I bylo dosaženo u 41,9 % pacientů s RUXO redukce velikosti sleziny ≥ 35 % ve 24. týdnu vs. 0,7 % ve větvi s placebem. Podobný efekt byl pozorován i na redukci celkových příznaků (45,9 % vs. 5,3 %). Ve studii COMFORT II dosáhlo redukce velikosti sleziny ≥ 35 % ve 24. týdnu 32 % vs. 0 % pacientů z placebové větve, ve 48. týdnu to bylo 28 % vs. 0 % pacientů. Z výsledků studií je také potvrzeno, že dosažení léčebného efektu není závislé na přítomnosti mutace JAK2 [15].
Výsledky obou studií potvrdily, že zahájení léčby RUXO u pacientů zlepšuje celkové přežití (overall survival, OS). Při pětileté analýze dat byla zjištěna 30% redukce úmrtí u nemocných randomizovaných do větve s RUXO, kdy OS ve větvi s RUXO je 5,3 roku vs. 3,8 roku v kontrolní skupině. V těchto studiích byla u 60 % pacientů zjištěna minimálně 50% redukce palpovatelné sleziny. Význam redukce velikosti sleziny pro OS či progresi choroby u pacientů s PMF je stále předmětem diskusí. Je ale prokázáno, že OS u pacientů se zmenšením sleziny alespoň o 25 % bylo delší než u pacientů beze změny splenomegalie při léčbě. Ve studiích COMFORT bylo rovněž zjištěno, že výhoda přežití u pacientů léčených RUXO není závislá na anémii a na potřebě transfuzí [15].
Velkým přínosem léčby RUXO pro pacienty s PMF je také rychlá kontrola symptomů choroby (pocení, hubnutí, únava), ke které dochází zablokováním dráhy JAK-STAT s následným poklesem hodnot celé řady prozánětlivých cytokinů. Pro pacienta bývá tento faktor ovlivňující kvalitu života často rozhodujícím.
Nežádoucí účinky léčby
ruxolitinibem
Negativem léčby RUXO je skutečnost, že po pěti letech terapie zůstává léčeno pouze 27 % pacientů, nejčastějšími důvody ukončení léčby jsou nežádoucí účinky (35 %) a progrese choroby (32 %). K nejčastějším nežádoucím účinkům řadíme hematologickou toxicitu ve smyslu anémie a trombocytopenie. Ve studiích se pohyboval výskyt anémie stupně 3 a 4 kolem 40−45 %, ale velká část případů byla reverzibilních. Výskyt trombocytopenie stupně 3 a 4 je kolem 10–12 % případů s nutností přerušení či úpravy dávky RUXO [16,17].
Jak zmírnit anémii při léčbě RUXO, není stále jasné. U mladších pacientů, kteří výhledově budou indikováni k HSCT, je možností přidání chelatační léčby nebo můžeme léčbu rozšířit o erytropoézu stimulující látky (r EPO). V současné době rovněž probíhají studie, kde se do kombinace s RUXO přidává sotatercept nebo luspatercept, které se váží na ligandy TGFβ (transforming growth factor beta) a zlepší vyzrávání erytrocytárních prekurzorů (tab. 1).
K dalším častým nežádoucím
účinkům patří infekční komplikace. Výskyt infekcí je
přítomen u 10−20 % pacientů léčených RUXO a výskyt
narůstá s pokročilostí choroby a s velikostí
splenomegalie. K velmi častým patří infekce močové
a plicní. Relativně často se objevují infekce herpetického
původu, kde u rizikových pacientů je vhodné při zahájení
léčby RUXO začít podávat i antivirovou profylaxi. Byly
rovněž popsány případy reaktivace hepatitid a několik
případů rozvoje tuberkulózy či progresivní multifokální
leukoencefalopatie. Pacienti léčení RUXO by měli být po stránce
možné infekční komplikace pečlivě sledováni [18−21].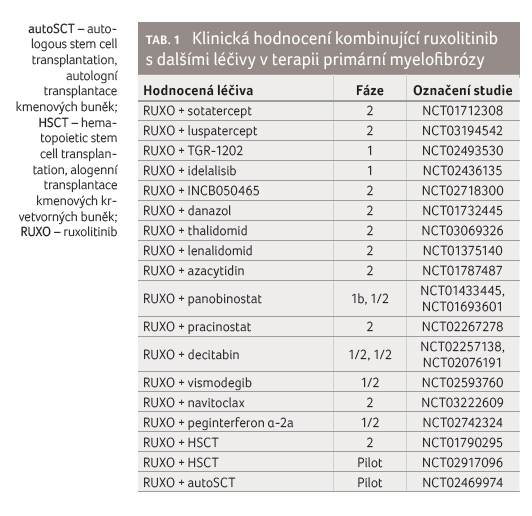
Podle výstupu studií COMFORT I a COMFORT II je u pacientů léčených RUXO zvýšený výskyt kožních nemelanomových nádorů, a proto je nutno věnovat pozornost i této oblasti. Na druhou stranu z několika předchozích studií vyplývá, že pacienti s MPN mají asi 2−3× vyšší riziko dalších malignit. Tento typ kožního nádoru se často objevuje i při léčbě hydroxyureou, která nezřídka léčbu RUXO předchází [16].
Ruxolitinib u PV a ET
Schválení RUXO do léčby PV umožnily výsledky studie RESPONSE, ve které pacienti s PV rezistentní/intolerantní k hydroxyuree byli randomizováni do větve s RUXO či do větve s BAT. Při hodnocení efektu léčby ve 32. týdnu došlo ke kontrole hematokritu u 60 % pacientů léčených RUXO proti 19,6 % pacientů ve větvi s BAT; zmenšení velikosti sleziny alespoň o 35 % bylo docíleno u 38,2 % pacientů ve skupině RUXO vs. u 0,9 % pacientů ve skupině BAT a kompletní hematologické odpovědi bylo dosaženo u 23,6 % pacientů ve skupině RUXO oproti 8,9 % pacientů ve skupině BAT. U téměř poloviny pacientů ve větvi s RUXO došlo rovněž k významné redukci symptomů choroby hodnocené pomocí MPD dotazníku (49 % vs. 5 %), a tedy ke zlepšení kvality života [22].
Velmi pozitivním zjištěním při hodnocení výstupů této studie je redukce výskytu trombotických příhod u pacientů léčených RUXO. Při hodnocení výsledků v 80. měsíci léčby byl jejich výskyt 1,8 × 100 pacientoroků vs. 8,2 × 100 pacientoroků (RUXO vs. BAT) a při čtyřleté analýze došlo k další redukci výskytu trombóz na 1,2 × 100 pacientoroků. Zajímavá bude rovněž odpověď na otázku, zda zahájení léčby RUXO u pacientů s PV sníží frekvenci progrese choroby do postpolycytemické PMF či sekundární leukemie [22].
Zatím ne zcela přesvědčivé výsledky podávání RUXO byly prokázány při jeho užití v léčbě pacientů trpících ET. Ve studii MAJIC ET bylo při hodnocení po jednom roce dosaženo kompletní odpovědi u 46,6 % pacientů léčených RUXO a u 44,2 % léčených jinak. Po dvou letech léčby byl rovněž srovnatelný počet transformací choroby a výskyt trombotických či krvácivých komplikací [23].
Indikace ruxolitinibu u pacientů
s PMF a PV
Ruxolitinib (Jakavi) zatím nemá v ČR pro léčbu PMF a PV úhradu a je třeba o ni žádat v souladu s §16 zákona č. 48/1997 Sb., v platném znění. U PMF je RUXO indikován k léčbě dospělých pacientů se splenomegalií nebo s příznaky přidruženými k PMF. U pacientů trpících PV patří RUXO do skupiny 2. linie léčby pro nemocné, kteří jsou rezistentní či intolerantní k předchozí léčbě hydroxyureou.
Dávkování přípravku u PMF se řídí vstupními hodnotami trombocytů, dávka 15 mg podávaná dvakrát denně je určena pacientům s počtem trombocytů 100−200 × 109/l a dávka 20 mg dvakrát denně pro pacienty s počtem trombocytů > 200 × 109/l. U pacientů s počtem trombocytů 50–99 × 109/l je doporučena úvodní dávka 5 mg 2× denně za průběžné kontroly vývoje hodnot destiček.
U pacientů s PV je
doporučena úvodní dávka 10 mg perorálně 2× denně.
U pacientů s počtem trombocytů 50−99 × 109/l
je maximální doporučená úvodní dávka 5 mg 2× denně
a její další titrace má být prováděna velmi opatrně.
Pokud léčba není účinná a hodnoty trombocytů se nacházejí
v mezích normy, je možné navýšení dávky RUXO na 25 mg
2× denně.
Závěr
Ruxolitinib je prvním a stále jediným přípravkem ze skupiny inhibitorů JAK1/2 schváleným pro léčbu primární myelofibrózy a onemocnění polycythaemia vera. V současné době máme již dostatečné množství informací o jeho účinnosti, bezpečnosti a pozitivním ovlivnění přežití nemocných trpících PMF. Pacienti především příznivě hodnotí významné zlepšení kvality života. U vybrané skupiny pacientů s PV představuje pro nás další léčebnou možnost kontroly hematokritu se současně příznivou redukcí výskytu trombotických komplikací [24−26].
U pacientů s PMF při
selhání léčby RUXO nemáme k dispozici mnoho dalších
léčebných alternativ. Od schválení RUXO již uběhlo sedm
let, která však překvapivě nepřinesla do klinické praxe
nové přípravky ze skupiny inhibitorů JAK1/2 (tab. 2).
Bohužel léčba PMF inhibitory JAK1/2 se tak nestala analogií léčby
chronické myeloidní leukemie tyrozinkinázovými inhibitory. Možnou
budoucnost léčby představují kombinace RUXO s přípravky
z jiných skupin, které by pozitivně ovlivnily nežádoucí
účinky RUXO, jako je např. anémie, a rovněž by posílily
léčebnou odpověď RUXO (tab. 1).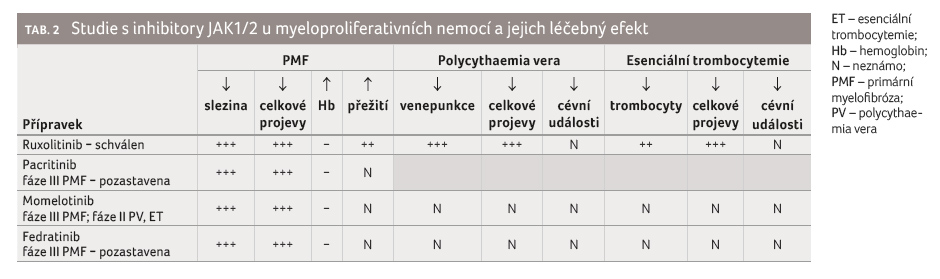
Seznam použité literatury
- [1] Tefferi A, Lasho TL, Steensma DP, et al. The JAK2V617F tyrosine kinase mutation in myelofibrosis with myeloid metaplasia: lineage specifity and clinical correlates. Br J Haematol 2005; 131: 320–328.
- [2] Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med 2013; 19; 2391–2405.
- [3] Tefferi A, Lasho TL, Finke CM, et al. CALR vs JAK2 vs MPL‑mutated or triple‑negative myelofibrosis: clinical, cytogenetic and molecular comparisons. Leukemia 2014; 28: 1472–1477.
- [4] Titmarsh GJ, Duncombe AS, McMullin MF, et al. How common are myeloproliferative neoplasms? A systematic review and meta‑analysis. Am J Hematol 2014; 89: 581–587.
- [5] Tefferi A, Guglielmelli P, Larson DR, et al. Long‑term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014; 124: 2507–2513.
- [6] Tefferi A, Rumi E, Finazzi G, et al. Survival and prognosis among 1545 patients with contemporary polycythemia vera: an international study. Leukemia 2013; 27: 1874–1881.
- [7] Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood 2009; 113: 2895–2901.
- [8] Passamonti F, Cervantes F, Vannucchi AM, et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG‑MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood 2010; 115: 1703–1708.
- [9] Gangat N, Caramazza D, Vaidya R, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol 2011; 29: 392–397.
- [10] Mesa RA, Jamieson C, Bhatia R, et al. NCCN Guidelines Insights: Myeloproliferative Neoplasms, Version 2.2018. J Natl Compr Canc Netw 2017; 15: 1193–1207.
- [11] Vannucchi AM, Harrison CN. Emerging treatments for classical myeloproliferative neoplasms. Blood 2017; 129: 693–703.
- [12] Tefferi A, Mesa RA, Nagomey DN, et al. Splenectomy in myelofibrosis with myeloid metaplasia: a single‑institution experience with 223 patients. Blood 2000; 95: 2226–2233.
- [13] Tefferi A, Jimenez T, Gray LA, et al. Radiation therapy for symptomatic hepatomegaly in myelofibrosis with myeloid metaplasia. Eur J Hematol 2001; 66: 37–42.
- [14] O‘Sullivan JM, Harrison CN. JAK‑STAT signaling in the therapeutic landscape of myeloproliferative neoplasms. Mol Cell Endocrinol 2017; 451: 71–79.
- [15] Verstovsek S, Mesa RA, Gotlib J, et al. Efficacy, safety, and survival with ruxolitinib in patients with myelofibrosis: results of a median 3‑year follow‑up of COMFORT‑I. Haematologica 2015; 100: 479–488.
- [16] Verstovsek S, Mesa RA, Gotlib J, et al. Long‑term treatment with ruxolitinib for patients with myelofibrosis: 5‑year update from the randomized, double‑blind, placebo‑controlled, phase 3 COMFORT‑I trial. J Hematol Oncol 2017; 22:10: 55.
- [17] Pardanani A, Tefferi A. Definition and management of ruxolitinib treatment failure in myelofibrosis. Blood Cancer J 2014; 4: 1–7.
- [18] Lussana F, Cattaneo M, Rambaldi A, Squizzato A. Ruxolitinib‑associated infections: A systematic review and meta‑analysis. Am J Hematol 2018; 93: 339–347.
- [19] Hopman RK, Lawrence SJ, Oh ST. Disseminated tuberculosis associated with ruxolitinib. Leukemia 2014; 28: 1750–1751.
- [20] Tong LX, Jackson J, Kerstetter J, Worswick SD. Reactivation of herpes simplex virus infection in a patient undergoing ruxolitinib treatment. J Am Acad Dermatol 2014; 70: 59–60.
- [21] Lee SC, Feenstra J, Georghiou PR. Pneumocystis jiroveci pneumonitis complicating ruxolitinib therapy. BMJ Case Rep 2014; 2014: doi: 10.1136/bcr‑2014‑204950.
- [22] Griesshammer M, Saydam G, Palandri F, et al. Ruxolitinib for the treatment of inadequately controlled polycythemia vera without splenomegaly: 80‑week follow‑up from the RESPONSE‑2 trial. Ann Hematol 2018; 97: 1591–1600.
- [23] Harrison CN, Mead AJ, Panchal A, et al. Ruxolitinib vs best available therapy for ET intolerant or resistant to hydroxycarbamid. Blood 2017; 130: 1889–1897.
- [24] Bryan JC, Verstovsek S. Overcoming treatment challenges in myelofibrosis and polycythemia vera: the role of ruxolitinib. Cancer Chemother Pharmacol 2016; 77: 1125–1142.
- [25] Bose P, Verstovsek S. JAK2 inhibitors for myeloproliferative neoplasms: what is next. Blood 2017; 130: 115–125.
- [26] Passamonti F, Maffioli M. The role of JAK2 inhibitors in MPNs 7 years after approval. Blood 2018; 131: 2426–2435.