Progresivní selhání β-buněk při diabetu mellitu 2. typu a možnosti léčebného ovlivnění
Rozvoj inzulinového deficitu vycházejícího z progredující apoptózy b-buněk je základním rysem diabetu 2. typu. Hlavním patogenetickým dějem je zvýšený oxidační stres, který indukuje stres endoplazmatického retikula. Převaha proapoptotických proteinů skupiny Bcl-2 vede následně k zániku mitochondrií, což je pak signálem pro zánik b-buňky. Farmakoterapie diabetu 2. typu by měla vycházet z patofyziologických poznatků, a tudíž využívat ty preparáty, které působí ochranně (antiapoptoticky) na b-buňku v kombinaci s léky snižujícími inzulinovou rezistenci.
Za základ rozvoje onemocnění diabetem mellitem 1. i 2. typu se považuje nedostatek inzulinu. Jde o poruchu, která podmiňuje nejen změny v glykoregulaci, ale i abnormality v metabolismu dalších základních živin, tedy tuků a proteinů. Stanovení glykemie jako jednoduchého indikátoru umožňuje do jisté míry kvantifikovat poruchu, i když k posouzení skutečného stupně nedostatku inzulinu je zapotřebí znát sekreci tohoto hormonu a zhodnotit též jeho periferní působení. Při definici diabetu se často používá označení „absolutní“ nebo „relativní“ nedostatek inzulinu, což kromě posouzení stupně poruchy zahrnuje i klinický aspekt, totiž požadavek na léčbu exogenním inzulinem. Pro správné rozhodnutí o volbě a vedení léčby diabetu je vhodná znalost fyziologických regulačních mechanismů a dále i posloupnost změn, které vyúsťují až do manifestního diabetu.
Sekrece a působení inzulinu za fyziologických podmínek
Sekrece inzulinu je u zdravého jedince v rovnováze s jeho působením v cílových tkáních. U osob s velkou citlivostí na inzulin, tzv. inzulin-senzitivních, je přítomna nízká sekrece inzulinu, neboť potřeba inzulinu k udržení glykemie ve fyziologickém rozpětí je malá [1]. Pokud by se tento vztah porušil a sekrece inzulinu by nepřiměřeně vzrostla, dostavily by se hypoglykemie. Tento stav se vyskytuje typicky u inzulinomu, u něhož autonomní sekrece inzulinu nepo-dléhá zpětné kontrole výše glykemie a náhlé uvolnění inzulinu zβ-buněkβ nádoru není vyrovnáno rezistencí tkání k inzulinu, čímž dochází k těžké hypoglykemii.
Jakmile se změní působení inzulinu v játrech, tukové tkáni či v příčně pruhovaném svalu, dojde i ke změně sekrece inzulinu. Vzestup inzulinové rezistence v játrech a periferních tkáních znamená i potřebu vyšší sekrece inzulinu, má-li se zachovat homeostáza glukózy a tím i normální hodnoty glykemií [1]. Naproti tomu pokles rezis-tence provází pokles sekrece inzulinu. Zvýšení inzulinové rezistence se typicky vyskytuje při nárůstu hmotnosti při vyšším energetickém příjmu nebo při snížené fyzické aktivitě, což způsobuje pozitivní energetickou bilanci, a tedy akumulaci energie. Naopak pokles hmotnosti navozený intenzivním redukčním režimem nebo zvýšenou fyzickou aktivitou se projeví poklesem sekrece inzulinu. Molekulární podstata vztahu uvedených regulací se-krece a působení inzulinu nebyla dosud zcela objasněna, i když již dříve se uvažovalo o přímém vztahu mezi inzulinovým receptorem v cílové tkáni a sekrecí inzulinu z β-buňky.
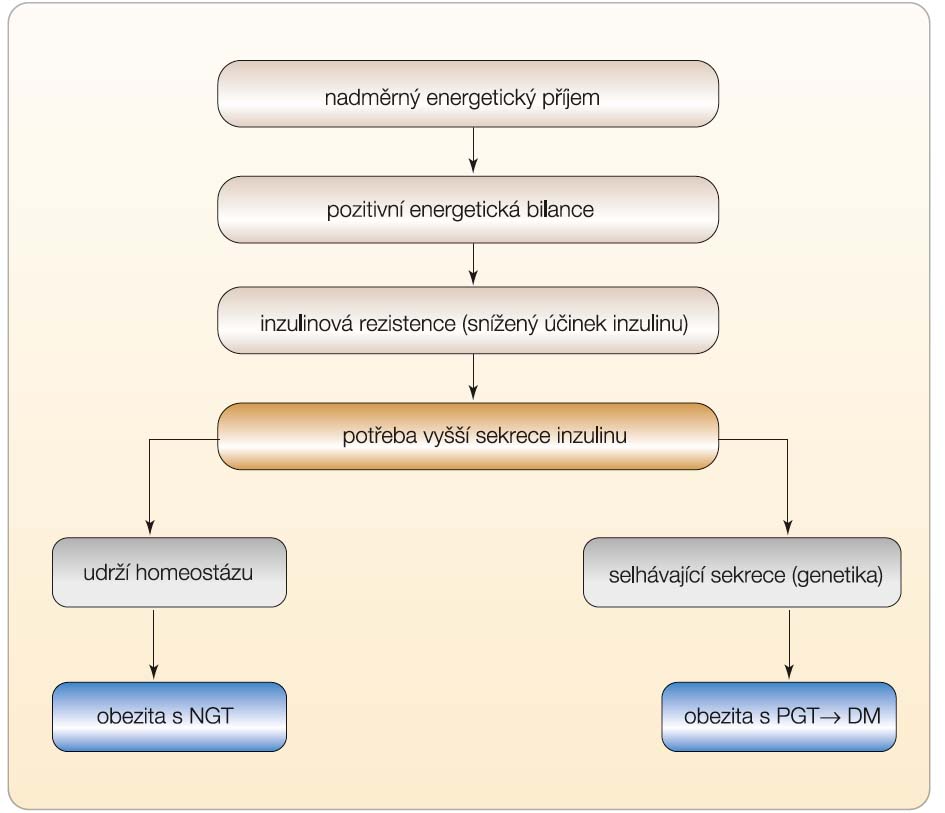 Chronicky zvýšená inzulinová rezistence podmiňuje trvale zvýšenou sekreci inzulinu, která vede k hyperinzulinemii. Tato situace je běžná u obézních osob, jejichž bazální koncentrace inzulinu v plazmě nalačno je několikanásobně vyšší v porovnání s neobézními inzulin-senzitivními jedinci. Pokud má jedinec dostatečnou kapacituβ-buněk k sekreci inzulinu a nemá-li genetickou predispozici k jejich selhání, zůstane zachována normoglykemie navzdory velké rezistenci na inzulin (obr. 1). Histologicky je přítomna u těchto osob hyper-trofie ostrůvků se zmnoženímβ-buněk [2]. Hyperinzulinemie trvající i postprandiálně může někdy vyvolat i mírný pokles glykemie s pocitem hladu a potřebou konzumace jídla, takže vede v některých případech i k podezření, že se jedná o organickou příčinu, tj. například o inzulinom. To nastává zejména v situaci, kdy jedinec zkonzumuje jen sladký pokrm a stimulace inzulinu dočasně „přestřelí“ skutečnou potřebu organismu. Správné vyhodnocení situace však umožní navrhnout vhodné doporučení, především úpravu dietních návyků.
Chronicky zvýšená inzulinová rezistence podmiňuje trvale zvýšenou sekreci inzulinu, která vede k hyperinzulinemii. Tato situace je běžná u obézních osob, jejichž bazální koncentrace inzulinu v plazmě nalačno je několikanásobně vyšší v porovnání s neobézními inzulin-senzitivními jedinci. Pokud má jedinec dostatečnou kapacituβ-buněk k sekreci inzulinu a nemá-li genetickou predispozici k jejich selhání, zůstane zachována normoglykemie navzdory velké rezistenci na inzulin (obr. 1). Histologicky je přítomna u těchto osob hyper-trofie ostrůvků se zmnoženímβ-buněk [2]. Hyperinzulinemie trvající i postprandiálně může někdy vyvolat i mírný pokles glykemie s pocitem hladu a potřebou konzumace jídla, takže vede v některých případech i k podezření, že se jedná o organickou příčinu, tj. například o inzulinom. To nastává zejména v situaci, kdy jedinec zkonzumuje jen sladký pokrm a stimulace inzulinu dočasně „přestřelí“ skutečnou potřebu organismu. Správné vyhodnocení situace však umožní navrhnout vhodné doporučení, především úpravu dietních návyků.
Cesta k inzulinové deficienci – patogenetické a patofyziologické aspekty
U jedince s genetickou predispozicí k diabetu navodí dlouhodobě pozitivní energetická bilance inzulinovou rezistenci, kterou však β-buňky nejsou schopny dlouhodobě vyrovnávat (obr. 1). Zvýšená energetická nálož v podobě příjmu nasycených tuků, ale i sacharidů zvyšuje nároky na sekreci inzulinu podmíněnou zejména volnými mastnými kyselinami (FFA). Uplatní se lipotoxický a později, po vzestupu glykemie, i glukotoxický efekt, souborně označovaný též jako glukolipotoxický, který při současné přítomnosti kandidátních genů vede k selhání sekrece potřebného množství inzulinu. Tento děj je později provázen úbytkem hmotyβ-buněk spolu se selhávající sekreční kapacitou a morfologickým průkazem apoptózy, která probíhá rychleji, než by odpovídalo regeneraciβ-buněk. Rychlost rozvoje diabetu je ovlivněna souhrou genetické predispozice a tíží metabolické poruchy podmíněné nároky na sekreci inzulinu při stoupající inzulinové rezistenci, která proces apoptózy zesiluje.
Hlavním podnětem pro zahájení apoptózy β-buněk jsou volné mastné kyseliny (FFA), zejména kyselina palmitová, a dále pak hyperglykemie, jakmile se začne vytvářet. U jedince s normoglykemií je prvním stimulem dlouhodobě zvýšená plazmatická koncentrace FFA, která ovlivní β-buňku jevem označovaným jako lipotoxicita. V současné době se předpokládají tři mechanismy, jimiž se uplatňuje působení FFA na β-buňku [3]. Zaprvé FFA indukují stres endoplazmatického retikula (ER) vystupňovaným procesem jejich esterifikace, zadruhé FFA indukují tvorbu oxidačního stresu v β-buňce a zatřetí zvyšují tvorbu ceramidu z palmitátu, který působí proapoptoticky a snižuje genovou ex-presi inzulinu. Významná je z hlediska lipotoxicity dlouhodobě zvýšená koncentrace FFA, kdežto její krátkodobý vzestup působí na β-buňku stimulačně a zvyšuje sekreci inzulinu [4]. Podobně se projevuje vyšší koncentrace glukózy, která se více trans-portuje do β-buňky a stimuluje sekreci inzulinu. Dlouhodobá expozice zvýšené koncentraci glukózy však způsobuje de-senzitizaci β-buňky, která přestává být na glukózu dostatečně citlivá. V β-buňce podmiňuje metabolismus glukózy tvorbu malonyl-koenzymu A, jehož oxidace v mitochondriích zabraňuje oxidaci volných mastných kyselin, které se pak v podobě esterů hromadí v cytoplazmě. Touto cestou oba substráty (FFA a glukóza) negativně ovlivňují jak svůj metabolismus, tak i děje související s funkcí organel.
![Kaskáda reakcí vedoucích k apoptóze β-buňky indukovaná palmitátem při diabetu 2. typu; podle [6] – Gurzov, et al. ER – endoplazmatické retikulum, ATF3 – regulační molekula, DP5 – protein, Mcl-1 – antiapoptotický protein, Bcl-XL – antiapoptotický protein, PUMA – p53-upregulated modulator of apoptosis, Bax – proapoptotický protein](https://www.remedia.cz/photo-a-28822---.jpg) Palmitátem podmíněný stres endoplazmatického retikula vede k potlačení anti-apoptotických Bcl-2 proteinů Mcl-1 a Bcl-XL, jež chrání buňku před apoptózou, a současně se zvyšuje exprese proteinu DP5, kterou podmiňují regulační molekuly JNK a ATF3 [5] (obr. 2). Celý komplex dějů vedoucí k apoptóze β-buňky byl nedávno popsán a byly porovnány rozdíly mezi oběma typy diabetu [6]. V experimentálních studiích se potvrdilo, že stimulace ochranných Bcl-2 proteinů, tj. Mcl-1 a Bcl-XL, může zabránit apoptóze β-buněk [7, 8], ale tím se neovlivní porucha sekrece inzulinu vyvolaná cytokiny. Znamená to, že oba děje, tj. zánik β-buněk a jejich funkce, jsou odděleně řízeny.
Palmitátem podmíněný stres endoplazmatického retikula vede k potlačení anti-apoptotických Bcl-2 proteinů Mcl-1 a Bcl-XL, jež chrání buňku před apoptózou, a současně se zvyšuje exprese proteinu DP5, kterou podmiňují regulační molekuly JNK a ATF3 [5] (obr. 2). Celý komplex dějů vedoucí k apoptóze β-buňky byl nedávno popsán a byly porovnány rozdíly mezi oběma typy diabetu [6]. V experimentálních studiích se potvrdilo, že stimulace ochranných Bcl-2 proteinů, tj. Mcl-1 a Bcl-XL, může zabránit apoptóze β-buněk [7, 8], ale tím se neovlivní porucha sekrece inzulinu vyvolaná cytokiny. Znamená to, že oba děje, tj. zánik β-buněk a jejich funkce, jsou odděleně řízeny.
V komplexním procesu apoptózy sehrává významnou úlohu vzájemná interakce endoplazmatického retikula a mitochondrií [9]. Rozvoj stresu endoplazmatického retikula je nezbytnou součástí celého procesu apoptózy, neboť jeho modulačním účinkem jsou potlačeny ochranné Bcl-2 proteiny Mcl-1 a Bcl-XL a naopak aktivovány proteiny DP5 a PUMA prostřednictvím dalších proteinových faktorů, které způsobují selhání mitochondrií. Na molekulární úrovni se na procesu vzájemné interakce ER a mitochondrií podílejí iontové kanály, tedy kalciový kanál a KATP kanál, jejichž normální funkce zajišťuje integritu a fyziologické fungování obou organel [10]. Chronicky zvýšená cytoplazmatická koncentrace kalcia, k níž může docházet při delší expozici β-buňky glukóze, ale i FFA, stimuluje mitochondrie ke zvýšené tvorbě reaktivních forem kyslíku (ROS) a tím je vede i k větší zranitelnosti.
Na rozdíl od zdravých jedinců, u nichž dobrá genetická dispozice a současně vhodná životospráva způsobí, že expozice reaktivním formám kyslíku má pozitivní vliv na regulační děje, u jedinců s genetickou vlohou k diabetu vyvolá dlouhodobě zvýšená tvorba ROS v β-buňkách selhávání funkcí způsobené narušením jejich organel. ER i mitochondrie jsou citlivé na oxidační stres, k němuž v β-buňce poměrně velmi snadno dochází, neboť ochranné (antioxidační) mechanismy jsou v ní slabší než v jiných buňkách [3]. Jde především o antioxidačně působící scavengerové enzymy, jako je superoxiddismutáza (SOD), kataláza (CAT) a glutathionperoxidáza (GPx), jejichž kapacita a tím i antioxidační pů-sobení je omezené. Navíc mohou být inhibovány hyperglykemií, což dále snižuje jejich účinek.
Vyšší oxidační stres se vyznačuje zvýšenou produkcí ROS, která probíhá jednak v mitochondriích při zvýšené dodávce substrátů pro oxidativní fosforylaci, jednak aktivovanou NADPH oxidázou, která se podílí na dalším zesílení stupně stresu. U hlodavců byl prokázán vlivem vyšší koncentrace glukózy zvýšený obsah peroxidu uvnitř ostrůvků [11]. Zvýšení obsahu superoxidového radikálu a peroxidu vodíku uvnitř buněk urychluje selhání obou zmíněných organel, ER a mitochondrií.
Selhávání mitochondrií je nitrobuněčným signálem pro aktivaci hydrolytických enzymů označených jako kaspázy, které podmíní apoptózu β-buňky. Počáteční fáze tohoto procesu nejsou laboratorně ani klinicky v běžné praxi detekovatelné, neboť i pokles počtu a hmoty β-buněk např. o 20–30 % se ještě neprojeví v glykoregulaci, jedinec je normoglykemický, ale jeho glykemie nalačno začíná postupně stoupat v rámci normálního rozpětí.
Tento děj je možno pozorovat i fyziologicky při procesu stárnutí, u něhož úbytek hmoty β-buněk způsobuje pozvolný vzestup glykemie. Jakmile apoptóza dosáhne u daného jedince kritického stupně, který též souvisí s jeho inzulinovou rezistencí, objeví se porucha metabolismu glukózy, tedy hraniční glykemie nalačno, a dále porušená glukózová tolerance. Jedinec je stále asymptomatický a diagnóza je možná pouze na základě náhodně provedeného vyšetření.
Sekrece inzulinu se vlivem progredující apoptózy postupně snižuje a při dosažení určité kritické hodnoty (pokles pod 50 % fyziologického množství β-buněk) již nestačí k pokrytí potřeby pro zachování euglykemie. Organismus se tak dostává do stadia relativního nedostatku inzulinu. Při výrazné inzulinové rezistenci může sekrece být ještě natolik zachována, že u jedince bez této rezistence by stejná sekrece zcela udržela metabolickou rovnováhu. Toto stadium je stále ještě reverzibilní a záleží především na opatřeních, která by snížila potřebu inzulinu v organismu. Nejde tedy o absolutní deficit inzulinu. Tato relativní hypoinzulinemie se však prohlubuje přetrvávajícími zvýšenými nároky organismu. Apoptóza β-buněk je kontinuální proces a podobně kontinuálně narůstá i glykemie nalačno. Na základě konvenční dohody je pak stanoven stupeň poruchy glukózové homeostázy, tedy zda se jedná o prediabetes (glykemie nalačno > 5,6 mmol/l nebo postprandiální glykemie 7,8–11,0 mmol/l), nebo již o diabetes (glykemie nalačno ≥ 7,0 mmol/l). Platí Starlingův vztah mezi glykemií a sekrecí inzulinu. Od flekčního bodu, který představuje přibližně hodnota 7 mmol/l, se průběh křivky obrací a další zvyšování glykemie je provázeno snižující se sekrecí inzulinu.
Rychlost průběhu apoptózy β-buněk a tím progrese poruchy včetně rozvoje deficitu inzulinu je závislá zejména na genetické vloze podmíněné různou kombinací zúčastněných kandidátních genů, které mohou přispívat i k tíži inzulinové rezistence, ale jejichž výčet a mozaikové zastoupení u daného jedince neznáme. Z logiky procesu pak vyplývá, že původně relativní inzulinový deficit přechází v průběhu již diagnostikovaného diabetu do stadia, kdy množství β-buněk a jejich funkční kapacita syntetizovat inzulin natolik poklesly, že lze stav označit jako absolutní inzulinový deficit. V tom případě je zapotřebí k úpravě metabolické poruchy podat exogenní inzulin. Stav byl proto již dříve označován jako diabetes mellitus závislý na inzulinu, i když tento pojem byl spojován (a v dosavadním výkazu klasifikace nemocí je spojován) s diabetem 1. typu.
Diabetes mellitus 2. typu není při stanovení diagnózy absolutně inzulin-deficitní, sekrece hormonu je zachována, ale už nestačí k udržení normoglykemie. Mírný nárůst glykemie nad normální hodnoty však většinou nepodmiňuje klinickou symptomatologii, a proto bývá diagnóza pozdní. V tomto období je rychlost apoptózy závislá nejen na účinku inzulinové rezistence, ale rovněž na výši glykemie, neboť se postupně uplatňuje také toxický vliv glukózy (glukotoxicita), která celkový proces urychluje. Navíc se zvýšená koncentrace glukózy promítá do tvorby pokročilých glykačních produktů (AGE, advanced glycation end products), které působí inhibičně na syntézu a sekreci inzulinu a zřejmě se podílejí na samotné apoptóze β-buňky [12].
Pochopení těchto patofyziologických vztahů je důležité provedení kauzální terapie diabetu. Současně odůvodňuje, proč moderní diabetologie usiluje o dva cíle. Zaprvé si klade požadavek odhalovat dosud nepoznané diabetiky co nejdříve, resp. zaměřovat se na osoby se zvýšeným rizikem a provádět u nich screening diabetu či prediabetu včetně následných opatření, a zadruhé ihned od stanovení dia-gnózy doporučuje velmi intenzivně léčit diabetes s cílem dosáhnout požadované (cílové) hodnoty. Smyslem obou cílů je zpomalit apoptózu a tím i rozvoj inzulinového deficitu.
Terapeutické možnosti k ovlivnění inzulinového deficitu
Kontinuální vývoj a prohlubování inzu-linového deficitu vede k otázce, jak u diabetika postupovat v tomto vývoji terapeuticky. Vedle inzulinového deficitu je při diabetu mellitu 2. typu vždy přítomen určitý stupeň inzulinové rezistence. Je tudíž zřejmé, že jak snížení inzulinové rezistence, tak pokles glykemie (se snížením glukotoxického účinku) povede ke zpomalení apoptózy β-buněk, a tedy i ke zpomalení úbytku sekrece inzulinu. Z tohoto pohle-du je pochopitelné využití metforminu od počátku terapie diabetu 2. typu. Vzhledem k jeho dalším pozitivním účinkům je využití metforminu zdůvodněné i ve fázi hraničních poruch, tedy u prediabetu. Je ovšem samozřejmé, že součástí účinných opatření jsou i nefarmakologické postupy, tedy hypokalorická dieta u osob s nadváhou a u obézních pacientů s prediabetem a za-řazení pravidelné vhodné fyzické aktivity. Logicky patří do skupiny opatření snižujících inzulinovou rezistenci i glitazony, z nichž zatím nyní zůstává k dispozici jediný – pio-glitazon. Je proto nezbytné posoudit, nakolik je přítomna inzulinová rezistence, kterou se nedaří uspokojivě ovlivnit metforminem, a pak je možno přidat pioglitazon, nejsou-li zásadní kontraindikace k jeho podání.
Většina antidiabetik je zaměřena na snížení glykemie, v převážné míře ovlivněním zbývající sekrece inzulinu. Obecnou ne-výhodou tzv. inzulinových sekretagog je riziko rozvoje hypoglykemie ze stimulace sekrece inzulinu, a to podle účinku daného preparátu a bez ohledu na aktuální potřebu organismu. Tím dochází i k hmotnostnímu přírůstku následkem větší konzumace jídla indukované hypoglykemizujícím účinkem léčby. Mezi sekretagoga řadíme jednak deriváty sulfonylurey a jednak glinidy. Tyto látky působí cestou draslíkového kanálu (KATP) tak, že jeho uzavření je signálem pro otevření kalciového kanálu s následným vzestupem koncentrace nitrobuněčného kalcia, které pak způsobí exocytózu granulí obsahujících inzulin. Uvedená stimulace zejména nej-silnějším a dlouze působícím glibenklamidem nikterak nešetříβ-buňku, ale naopak ji může vyčerpávat. V experimentech pak byla pozorována zvýšená apoptóza [13]. Experimentální práce doložila, že apoptóza β-buněk je podmíněna SUR1 podjednotkou sulfonylureového receptoru, na niž se váže glibenklamid [14]. Proto v současné době, kdy přibývají poznatky o průběhu apoptózy, může být využití některých sekretagog zpochybňováno. Jistě se pak klade otázka, nakolik je jejich použití vhodné i se zřetelem k urychlenému selhání a potřebě léčby inzulinem. I mezi deriváty sulfonylurey jsou rozdíly, u některých byly pozorovány antioxidační účinky (např. u gliklazidu), které by mohly působit příznivě [15]. Proto se nyní uvádí duální působení derivátů sulfonylurey, které spočívá jednak ve stimulaci sekrece inzulinu, jednak v antioxidačním vlivu, jenž by se mohl promítat do stabilizace β-buňky, a tedy do antiapoptotického efektu [10]. K ověření tohoto působení však zatím chybějí dlouhodobější studie.
Z fyziologického hlediska se jeví jako vhodný kandidát zlepšující sekreci inzulinu ten, který nejen vhodně stimulujeβ-buňku, ale současně ji chrání a nevede k jejímu vyčerpání a působí i pozitivně regeneračně, tedy proti apoptóze. Do této skupiny patří látky s inkretinovými účinky (inkretinová mimetika), a sice agonisté GLP-1 receptorů a inhibitory DPP-4 (gliptiny), u nichž se předpokládá ochranný vliv na β-buňky. Přestože zatím nejsou k dispozici výsledky dlouhodobého podávání, ex-perimentální data podporují předpoklad jejich protektivního účinku na β-buňky [16]. Nové poznatky v této oblasti mohou především ovlivnit léčebná schémata, a tedy algoritmus terapie diabetu 2. typu.
Samostatnou kapitolou v terapii časné fáze diabetu 2. typu je léčba inzulinem. Vzhledem k jeho rozmanitým účinkům, které přesahují úpravu glykemie v užším slova smyslu, je zatím jeho použití naprosto jednoznačné u stavů, kde je při diagnóze diabetu zjištěna glykemie obvykle vyšší než 15 mmol/l, a zejména je-li současně stav provázen zřetelnými symptomy. V takovém případě může vést podání inzulinu k rychlé úpravě glykemií, kdežto následně bývá inzulin vystřídán perorálními antidiabetiky. Naproti tomu zahájení léčby inzulinem v časné fázi vývoje diabetu, kdy metabolická kompenzace je jen mírně vychýlená, zůstává stále předmětem diskusí [17]. Vychází se obvykle z představy, že přítomná nadváha či obezita se při terapii inzulinem ještě zhorší a s tím se dále prohloubí inzulinová rezistence. Rozhodnutí o zahájení léčby inzulinem v časné fázi vývoje diabetu je snadnější, pokud je přítomen počáteční úbytek hmotnosti nebo je jedinec neobézní. V takovém případě se pomýšlí více na vliv inzulinového deficitu a podání exogenního inzulinu se pak považuje za logicky volenou terapii. V porovnání s glibenklamidem však kombinace metforminu s inzulinem má dlouhodobější účinek [18].
V případě léčby inzulinem ve stadiu relativního nedostatku inzulinu se předpokládá ochranný vliv hormonu naβ-buňku s možností zpomalení apoptózy. Zahájení terapie inzulinem je tudíž závislé nejen na aktuálním stavu pacienta a jeho onemocnění, ale také na zkušenostech a zvyklostech ošetřujícího lékaře. Ani v literatuře neexistuje obecné doporučení a přístup různých škol se liší. V každém případě by se měla upřednostnit individualizovaná, a tedy na pacienta orientovaná léčba s cílem zlepšit jeho metabolickou kompenzaci, která současně nepovede k nežádoucím vedlejším účinkům, např. k hypoglykemiím.
Stoupající výskyt diabetu v populaci implikuje větší genetickou zátěž tímto onemocněním s možností mísení genetické predispozice, proto se v posledních letech začíná ve světě hovořit o tzv. diabetu smíšeném. Tím se může stávat hranice mezi oběma typy diabetu stále méně ostrou, na počátku často nelze typ diabetu ihned rozlišit. Z pohledu patogeneze onemocnění je tato situace pochopitelná, neboť sekvence dějů je až na vyvolávající (spouštěcí) příčinu u obou typů diabetu prakticky shodná. V takovém případě lze považovat za odlišující indikátor jedině rychlost progrese apoptózy β-buněk. Její citlivé vyhodnocení v rámci klinického obrazu pacienta pak může vést i k individuálně volené terapii, včetně terapie inzulinem. V tomto stadiu je pak důležitější volba terapie než snaha o přesnou klasifikaci diabetu.
Závěr
Rozvoj inzulinového deficitu je rozhodujícím znakem při vývoji diabetu, přičemž jeho rychlost je podmíněna rychlostí apo-ptózy β-buněk. Správné zhodnocení tohoto dynamického procesu pak ovlivňuje volbu terapie, při níž je vždy vhodné posoudit, zda organismus má ještě rezervu reverzibilních dějů (např. snížení inzulinové rezis-tence), jejichž uplatnění může výrazně zpomalit proces vývoje diabetu. Na základě nových poznatků v současné době dochází i k revizi dosavadního léčebného algoritmu farmakoterapie diabetu 2. typu. Jako logická se z hlediska účinku jednotlivých léčiv jeví sekvence zahájená metforminem s následným poměrně časným začátkem podávání inkretinových antidiabetik v situaci, kdy samotný metformin nevede k úspěšné kompenzaci. V případě větší inzulinové rezistence se jako zdůvodněná jeví následně i kombinace s pioglitazonem. Pokud se nedosáhne zřetelnějšího zlepšení kompenzace diabetu, pak by se měla revidovat použitá kombinace a místo inkretinových antidiabetik by bylo vhodnější v takovém případě zahájit časné podávání inzulinu. Volba typu inzulinu musí vycházet z individuálního zhodnocení účinku u daného jedince (tj. dlouze působící inzulin přidaný k metforminu, nebo naopak přidání prandiálního krátce působícího inzulinu). Právě farmakoterapie diabetu 2. typu je příkladem velmi potřebného individualizovaného přístupu v léčbě onemocnění.
Práce vznikla za podpory výzkumného záměru MSM0021620807.
Seznam použité literatury
- [1] Weyer C, Bogardus C, Mott DM, Pratley RE. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J Clin Invest 1999; 104: 787–794.
- [2] Chang-Chen KJ, Mullur R, Bernal-Mizrachi E. Beta-cell failure as a complication of diabetes. Rev Endocr Metab Disord 2008; 9: 329–343.
- [3] van Raalte DH, Diamant M. Glucolipotoxicity and beta cells in type 2 diabetes mellitus: target for durable therapy? Diabetes Res Clin Pract 2011; 93: S37–S46.
- [4] Škrha J. Biochemie a patofyziologie. Diabetes mellitus, eds. J. Škrha, Galén 2009; 33–76.
- [5] Skrha J. Patogeneze diabetes mellitus 1. a 2. typu v roce 2011 – jednotící model poruchy glykoregulace. Vnitr Lek 2011; 57: 949–953.
- [6] Gurzov EN, Eizirik DL. Bcl-2 proteins in diabetes: mitochondrial pathways of beta-cell death and dysfunction. Trends Cell Biol 2011; 21: 424–431.
- [7] Allagnat F, Cunha D, Moore F, et al. Mcl-1 down-
- [8] Zhou YP, Pena JC, Roe MW, et al. Overexpression of Bcl-x(L) in beta-cells prevents cell death but impairs mitochondrial signal for insulin secre-tion. Am J Physiol Endocrinol Metab 2000; 278: E340–351.
- [9] Rieusset J. Mitochondria and endoplasmic reticulum: mitochondria-endoplasmic reticulum interplay in type 2 diabetes pathophysiology. Int J Biochem Cell Biol 2011; 43: 1257–1262.
- [10] Drews G, Krippeit-Drews P, Düfer M. Oxidative stress and beta-cell dysfunction. Pflugers Arch 2010; 460: 703–718.
- [11] Robertson RP, Harmon JS. Diabetes, glucose toxicity, and oxidative stress: A case of double jeopardy for the pancreatic islet beta cell. Free Rad Biol Med 2006; 41: 177–184.
- [12] Shu T, Zhu Y, Wang H, et al. AGEs decrease insulin synthesis in pancreatic beta-cell by repressing Pdx-1 protein expression at the post-translational level. PLoS One 2011; 6: e18782.
- [13] Sawada F, Inoguchi T, Tsubouchi H, et al. Differential effect of sulfonylureas on production of reactive oxygen species and apoptosis in cultured pancreatic beta-cell line, MIN6. Metabolism 2008; 57: 1038–1045.
- [14] Hambrock A, de Oliveira Franz CB, Hiller S, Osswald H. Glibenclamide-induced apoptosis is specifically enhanced by expression of the sulfonyl-urea receptor isoform SUR1 but not by expression of SUR2B or the mutant SUR1(M1289T). J Pharmacol Exp Ther 2006; 316: 1031–1037.
- [15] Del Guerra S, Grupillo M, Masini M, et al. Gliclazide protects human islet beta-cells from apoptosis induced by intermittent high glucose. Diabetes Metab Res Rev 2007; 23: 234–238.
- [16] Farilla L, Hui H, Bertolotto C, et al. Glucagon-like peptide-1 promotes islet cell growth and inhibits apoptosis in Zucker diabetic rats. Endocrinology 2002; 143: 4397–4408.
- [17] Lebovitz HE. Insulin: potential negative consequences of early routine use in patients with type 2 diabetes. Diabetes Care 2011; 34: S225–S230.
- [18] Fonseca V, Gill J, Zhou R, Leahy J. An analysis