Vedolizumab v léčbě Crohnovy nemoci a ulcerózní kolitidy
Souhrn:
Vedolizumab je monoklonální antiintegrinová protilátka typu IgG1 určená k léčbě nemocných s ulcerózní kolitidou a Crohnovou chorobou. Podávání vedolizumabu v klinické praxi bylo před několika měsíci povoleno v zemích EU, během tohoto roku se očekává, že bude dostupný také v České republice. Provedený klinický výzkum nazvaný GEMINI I–III ukázal vysokou účinnost a bezpečnost vedolizumabu v léčbě nemocných se středně až vysoce aktivní Crohnovou chorobou a ulcerózní kolitidou. Výhodou léčby vedolizumabem je vysoká účinnost také u nemocných, u nichž selhala léčba cílená proti tumor nekrotizujícímu faktoru alfa nebo jejichž organismus přestal na léčbu těmito přípravky odpovídat. Předností vedolizumabu je setrvalý účinek, který v průběhu dlouhodobého podávání narůstá, minimální imunogenicita a velmi vysoká bezpečnost, která je dána selektivním účinkem na gastrointestinální trakt. Určitou nevýhodou může být relativně pomalý nástup protizánětlivého účinku, omezené působení na mimostřevní projevy idiopatických střevních zánětů a malý potenciál pro léčení perianální formy Crohnovy nemoci.
Key words: vedolizumab, Crohn’s disease, ulcerative colitis, biological therapy.
Summary:
Vedolizumab is a monoclonal anti intergrin IgG1 antibody, which has been approved for use in clinical practice for Crohn´s disease and ulcerative colitis patients in European Union a few months ago. This drug is expected to be available during this year in the Czech Republic as well. The clinical research project GEMINI I–III proved that vedolizumab was very effective in patients with moderate to severe ulcerative colitis and Crohn´s disease. The significant advantages of vedolizumab therapy are high efficacy in patients who failed to respond to anti TNF alpha therapy, either primarily or secondarily. This drug shows a sustained effect increasing during long term maintenance therapy, minimal immunogenicity, and excellent safety profile, related to its high selecti-vity for gastrointestinal tract. There are also some disadvantages of vedolizumab treatment, which include slow antiinflammatory therapeutic response, limited effect on extraintestinal manifestations, and little therapeutic potential for perianal Crohn´s disease.
V květnu 2014 bylo v zemích Evropské unie (EU) a ve Spojených státech amerických povoleno pro klinickou praxi podávání vedolizumabu (VDZ) – nové molekuly ze skupiny biologických léčiv. Jde o humanizovanou monoklonální protilátku typu IgG1 proti leukocytárnímu integrinovému receptoru α4β7, která je určena pro léčbu ulcerózní kolitidy a Crohnovy nemoci. V některých zemích EU (Rakousko, SRN) se VDZ užívá v klinické praxi již od podzimu 2014. Předpokládá se, že v průběhu letošního roku bude přípravek uvolněn také pro farmaceutický trh v České republice a léčba vedolizumabem bude k dispozici i pro české pacienty s idiopatickými střevními záněty (inflammatory bowel disease, IBD).
Biologická léčba Crohnovy nemoci a ulcerózní kolitidy dosud zahrnovala výhradně monoklonální protilátky proti solubilnímu i membránově vázanému tumor nekrotizujícímu faktoru alfa (TNFα). Vedolizumab má zcela odlišný mechanismus účinku, který spočívá v zabránění vstupu aktivovaných bílých krvinek z krevního řečiště skrze kapilární stěnu do stře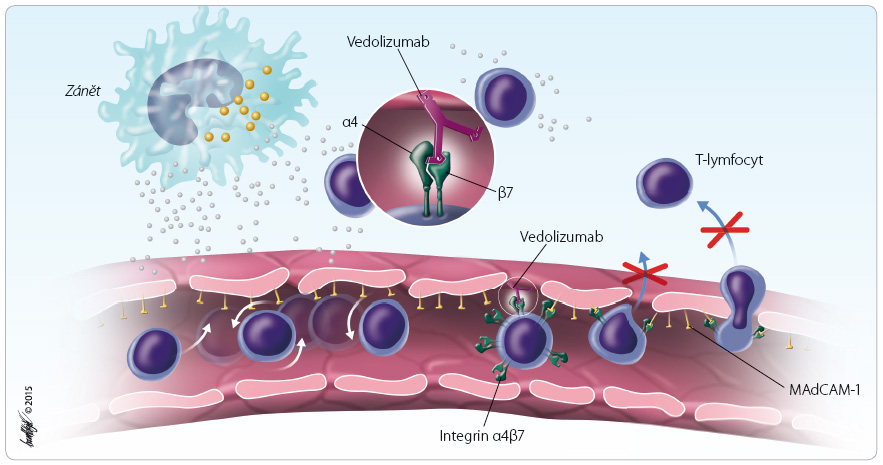 vní tkáně (obr. 1). Přilnutí aktivovaného leukocytu (T‑lymfocytu) se děje vazbou integrinového receptoru α4β7 na ligand MAdCAM‑1 (mucosal vascular addressin cell adhesion molecule 1), jenž je exprimován zejména na povrchu aktivovaných endoteliálních buněk kapilární sítě trávicího traktu.
vní tkáně (obr. 1). Přilnutí aktivovaného leukocytu (T‑lymfocytu) se děje vazbou integrinového receptoru α4β7 na ligand MAdCAM‑1 (mucosal vascular addressin cell adhesion molecule 1), jenž je exprimován zejména na povrchu aktivovaných endoteliálních buněk kapilární sítě trávicího traktu.
Nová generace tzv. antiintegrinových protilátek vykazuje významný posun především s ohledem na bezpečnost léčby, který je dán selektivním působením pouze na hybnost bílých krvinek v gastrointestinálním traktu [1]. Vývoj antiintegrinových protilátek probíhal od konce osmdesátých let minulého století a vedl k zavedení natalizumabu, monoklonální protilátky typu IgG4 cílené proti integrinové podjednotce α4. Vazbou na ni blokuje natalizumab nejen aktivitu integrinu α4β7 exprimovaného zejména ve střevě, ale také aktivitu integrinu α4β1, který je přítomen na povrchu leukocytů celého těla (vyjma neutrofilů). Neselektivní účinek natalizumabu je spojen s potenciálním rizikem neuroinfekce způsobené JC virem (John Cunningham virus, JCV) a vznikem progresivní multifokální leukoencefalopatie (PML). Riziko indukce této závažné neuroinfekce bylo hlavním důvodem toho, že v zemích EU, na rozdíl od Severní Ameriky, nebyl natalizumab povolen k léčbě pacientů s Crohnovou nemocí, ale pouze k léčbě nemocných s roztroušenou sklerózou [2,3].
Od roku 2006 probíhal s VDZ rozsáhlý klinický výzkum se zaměřením na jeho bezpečnost a účinnost u nemocných s ulcerózní kolitidou a Crohnovou nemocí. Projekt označený názvem GEMINI zahrnoval několik tisíc nemocných s IBD a proběhl ve třech stovkách center na pěti kontinentech. Česká republika se na tomto klinickém výzkumu podílela výraznou měrou a zařazením bezmála 140 pacientů s IBD patřila k důležitým partnerům projektu. Pozitivní výsledky klinického výzkumu byly podkladem k tomu, aby příslušné regulační orgány (Food and Drug Administration, FDA, a European Medicines Agency, EMA) povolily užívání VDZ v klinické praxi v USA i v zemích EU.
Vedolizumab v léčbě nemocných s ulcerózní kolitidou (GEMINI I)
GEMINI I byla randomizovaná, dvojitě zaslepená studie kontrolovaná placebem, která posuzovala účinnost a bezpečnost VDZ v indukční a udržovací léčbě nemocných s ulcerózní kolitidou se střední a vysokou aktivitou. Studie probíhala v letech 2008–2012 ve 211 výzkumných centrech 34 zemí. Podmínkou pro zařazení do studie bylo středně až vysoce aktivní onemocnění (skóre Mayo 6–12 bodů) s prokazatelným selháním léčebného účinku standardní léčby zahrnující glukokortikoidy a/nebo imunosupresiva nebo biologickou léčbu. Minimální rozsah nemoci byl stanoven na oblast rektosigmatu (≥ 15 cm). Složité uspořádání studie umožnilo postihnout oba parametry účinnosti: krátkodobý účinek hodnocený v týdnu 6 a dlouhodobé působení udržovací léčby v týdnu 52. Významnou součástí projektu bylo monitorování bezpečnosti a imunogenicity léčiva.
Do studie byli zahrnuti pacienti s ulcerózní kolitidou s tím, že endoskopické subskóre mělo hodnotu 2 nebo vyšší. Ve zdravotní dokumentaci každého zařazeného pacienta muselo být uvedeno selhání nebo závažné nežádoucí účinky dosavadní konvenční nebo biologické léčby. Koincidující terapie zahrnovala stabilní dávku mesalazinu, azathioprinu a/nebo glukokortikoidu (prednison ≤ 30 mg denně). V případě dosažení pozitivního účinku VDZ v indukční fázi studie (týden 6) bylo pacientům umožněno postupné ukončování léčby glukokortikoidy. V amerických centrech musela být ve stejné době ukončena terapie azathioprinem nebo 6‑merkaptopurinem, v ostatních „neamerických“ centrech byla imunosupresivní léčba ponechána po celou dobu studie [4].
Uspořádání studie GEMINI I
V incepční kohortě 1, která zahrnovala celkem 374 pacientů s ulcerózní kolitidou, bylo 225 nemocných randomizováno k podávání VDZ a 149 pacientům bylo podáváno placebo. Randomizace byla nastavena v poměru 3 : 2 ve prospěch VDZ v dávce 300 mg i.v. v týdnu 0 a v týdnu 2 se dvěma stratifikačními kritérii (současné užívání/neužívání glukokortikoidů a imunosupresiv a předchozí léčba antagonisty TNFα). Předcházející terapie přípravky anti‑TNFα byla omezena na 50 % pacientů zařazených do studie. Aby bylo dosaženo dostatečného počtu nemocných, kteří byli později převedeni do udržovací fáze studie, byla vytvořena ještě druhá kohorta (521 nemocných), kde indukce léčby VDZ probíhala dvěma infuzemi v týdnu 0 a 2 s hodnocením účinnosti v týdnu 6. Rozdíl proti první kohortě byl v tom, že všichni nemocní dostali účinnou látku, bez randomizace do ramene s placebem.
V šestém týdnu byla u všech pacientů zařazených do studie zhodnocena odpověď na podané dvě infuze VDZ. Pacienti, kteří dostávali VDZ a vykazovali klinickou odpověď v týdnu 6, byli opětovně randomizováni do udržovací fáze léčby k podávání placeba 1× za 8 týdnů (126 nemocných) nebo k podávání VDZ v dávce 300 mg 1× za 8 týdnů (122 pacientů), nebo k léčbě VDZ v dávce 300 mg 1× za 4 týdny (125 nemocných). V indukční fázi byl primárním cílovým ukazatelem podíl pacientů, kteří dosáhnou klinické odpovědi v týdnu 6, jež byla definována snížením hodnoty celkového skóre Mayo o 3 a více bodů a o 30 % celkové hodnoty skóre s tím, že hodnota subskóre pro krvácení mohla být 0 či 1. Sekundárním cílovým ukazatelem byl podíl pacientů, kteří dosáhli v týdnu 6 remise, jež byla definována hodnotou celkového skóre Mayo 2 a méně; slizniční zhojení hodnotou endoskopického subskóre 0 nebo 1.
Primárním cílovým ukazatelem pro udržovací fázi studie v týdnu 52 byl poměr pacientů v klinické remisi, sekundární cílové ukazatele studie zahrnovaly poměr nemocných s tzv. setrvalou klinickou odpovědí a remisí v období mezi 6. a 52. týdnem, podíl nemocných s dosaženým slizničním zhojením a poměr nemocných, kteří dosáhli remise bez léčby glukokortikoidy. Vedle parametrů účinnosti byla průběžně sledována také kvalita života hodnocená podle IBDQ (Inflammatory Bowel Disease Questionnaire) a trvale byl monitorován výskyt nežádoucích účinků. Do udržovací fáze studie bylo zařazeno celkem 373 pacientů.
Výsledky indukční fáze léčby (týden 6)
V týdnu 6 vykazovalo celkem 106 (47,1 %) ze 225 nemocných, jimž byly podány dvě infuze VDZ v týdnu 0 a 2, známky zlepšení v porovnání s placebovou skupinou, ve které splnilo kritéria klinické odpovědi jen 38 (25,5 %) ze 149 pacientů. Klinická remise byla zjištěna u 38 (16,9 %) nemocných, jimž byla podávána účinná látka, oproti 8 (5,4 %) nemocným, kteří dostávali placebo. Rozdíl mezi skupinami byl statisticky vysoce signifikantní (p = 0,001). Podíl nemocných, kteří dosáhli slizničního zhojení, byl v 6. týdnu při léčbě VDZ 92 (40,9 %) ze 225 léčených oproti 37 pacientům (24,8 %) ze 149 nemocných, jimž bylo podáváno placebo. Výsledky ve druhé „open‑label“ kohortě byly zcela srovnatelné s předchozími, 231 (44,3 %) pacientů z 521 léčených vykazovalo klinickou odpověď, 100 pacientů (19,2 %) se nacházelo v klinické remisi a 191 (36,7 %) dosáhlo slizničního zhojení.
Výsledky udržovací fáze léčby (týden 52)
V týdnu 52 byl počet remisí v aktivním rameni zjištěn u 51 (41,8 %) ze 122 nemocných, kteří byli léčeni VDZ podávaným po 8 týdnech, v porovnání s 56 (44,8 %) ze 125 pacientů, jimž byla podávána udržovací terapie po 4 týdnech. V rameni s placebem byl počet remisí významně nižší – 26 (15,9 %) ze 126 nemocných. Impresivní byly rozdíly mezi placebovou skupinou a aktivně léčenou skupinou dosahující 26,1 % v rameni VDZ s dávkováním po 8 týdnech (interval spolehlivosti – 95% CI: 14,9–37,2; p < 0,001) a 29,1 % v rameni s dávkováním VDZ po 4 týdnech (95% CI: 17,9–40,4; p < 0,001). Nebyl zaznamenán rozdíl v účinnosti v dosažení klinické remise a klinické odpovědi mezi oběma aktivními rameny s podáváním VDZ po 8 nebo po 4 týdnech. Analýza ukázala příznivý vliv VDZ na snížení parciálního indexu Mayo (symptomové zlepšení), snížení hodnoty fekálního kalprotektinu a zlepšení hodnot IBDQ v porovnání s placebem.
Naproti tomu nebyl zjištěn přídatný a pozitivní vliv konkomitantní terapie glukokortikoidy nebo imunosupresivy na celkový výsledek léčby VDZ. Příznivý výsledek byl zaznamenán v počtu dosažených remisí bez nutnosti podávání glukokortikoidů. V rameni, kde byly pacientům podávány infuze VDZ po 8 týdnech, byl poměr nemocných bez glukokortikoidů v 52. týdnu 31,4 %, u nemocných léčených VDZ po 4 týdnech byl poměr ještě vyšší – 45,2 %. V placebovém rameni dosáhl počet remisí bez podávání glukokortikoidů jen 13,9 %.
Monitorování bezpečnosti ukázalo, že výskyt nežádoucích účinků se mezi skupinou s aktivní léčbou a s placebem nelišil. Nebyl zaznamenán žádný případ PML ani žádné závažné infuzní reakce. V průběhu léčby nedošlo (na rozdíl od natalizumabu) ke zvyšování hodnoty celkového počtu lymfocytů v periferní krvi. Účinnost léčby korelovala s průměrnou sérovou koncentrací VDZ po dosažení rovnovážného stavu (11,2 ± 7,2 µg/ml při dávkování 1× za 8 týdnů vs. 38,3 ± 24,4 µg/ml při dávkování po 4 týdnech) [4,5].
Vedolizumab v léčbě nemocných s Crohnovou chorobou (GEMINI II a GEMINI III)
Studie GEMINI II
GEMINI II je randomizovaná, dvojitě zaslepená, placebem kontrolovaná klinická studie fáze III, která se uskutečnila v letech 2008 až 2012 ve 285 centrech 39 zemí. Do studie byli zařazeni nemocní se střední až vysokou aktivitou Crohnovy nemoci (Crohn´s disease activity index, CDAI 220–450) a s vyšší hodnotou C‑reaktivního proteinu, CRP (> 2,87 mg/l) nebo s vyšší hodnotou fekálního kalprotektinu (> 250 µg/g stolice) nebo s pozitivním nálezem vředů při kolonoskopickém vyšetření nebo při enterografickém vyšetření magnetickou rezonancí či počítačovou tomografií (MR/CT). Do studie byli zařazeni nemocní, kteří nevykazovali klinickou odpověď na terapii glukokortikoidy, imunosupresivy nebo na léčbu anti‑TNFα. Konkomitantní terapie byla stejná jako v předcházející studii GEMINI I [4].
Uspořádání studie GEMINI II
Uspořádání studie GEMINI II bylo totožné jako u výše uvedené studie GEMINI I. Indukční fáze byla hodnocena podle odpovědi pacientů v randomizované kohortě 1 kontrolované placebem. Vedolizumab v dávce 300 mg byl aplikován v týdnech 0 a 2 a v týdnu 6 byla zhodnocena odpověď na terapii. Randomizace byla stratifikována podle předcházející léčby glukokortikoidy, imunosupresivy, biologickou léčbou nebo jejich kombinací. Podíl nemocných s předcházející léčbou anti‑TNFα byl omezen maximálně na 50 % ze všech zařazených do klinického hodnocení. Aby byl počet hodnocených pacientů v udržovací fázi studie dostatečný, byla pro indukční léčbu otevřena ještě kohorta 2, v níž byl nemocným VDZ podáván v režimu „open label“ tj. všichni dostali účinnou látku v týdnech 0 a 2.
Jako pozitivní klinická odpověď bylo hodnoceno snížení indexu CDAI o 70 a více bodů v týdnu 6 v porovnání s výchozím stavem. Pacienti s příznivou odpovědí na indukční léčbu byli z obou kohort randomizováni do udržovací fáze studie v poměru 1 : 1 : 1, tj. VDZ 1× za 8 týdnů, VDZ 1× za 4 týdny nebo k podávání placeba až do týdne 52. Nemocní, kteří nedosáhli klinické odpovědi v týdnu 6, dostávali dále VDZ po 4 týdnech, a to až do konce studie. Dávka glukokortikoidů mohla být snižována a posléze léčba glukokortikoidy ukončena až od týdne 6; imunosupresiva mohla být v léčbě ponechána po celou dobu studie, vyjma amerických center, kde bylo nutné podávání imunosupresiv po týdnu 6 ukončit.
Výsledky indukční fáze studie (týden 6)
Indukční část studie měla dva primární cíle – určit podíl nemocných s významnou klinickou odpovědí (snížení indexu CDAI o více než 100 bodů) a dosažení klinické remise (celková hodnota CDAI ≤ 150 bodů) v týdnu 6. Do indukční fáze bylo randomizováno celkem 368 pacientů z kohorty 1. V týdnu 6 po dvou infuzích VDZ dosáhlo klinické remise celkem 32 (14,5 %) nemocných z 220 léčených oproti 10 pacientům (6,8 %) ze 148 ve skupině s placebem. Rozdíl ve výsledcích obou skupin byl statisticky signifikantní (p = 0,02). Významná klinická odpověď byla zaznamenána u 68 nemocných (31,4 %) léčených VDZ a u 38 pacientů (25,7 %) ze skupiny s placebem (p = 0,23); rozdíly v obou skupinách však nedosáhly statistické významnosti.
Výsledky udržovací fáze studie (týden 52)
Primárním cílovým ukazatelem udržovací fáze studie byl poměr nemocných s dosaženou klinickou remisí v týdnu 52; sekundárními cíli bylo zjistit počet nemocných s významnou klinickou odpovědí, podíl nemocných v remisi bez léčby glukokortikoidy a podíl nemocných s dosaženou setrvalou klinickou remisí, která byla definována výskytem klinické remise u více než 80 % kontrol v průběhu studie. Průběžně byl zaznamenáván výskyt nežádoucích účinků a vedlejších projevů léčby. Do udržovací fáze bylo randomizováno celkem 461 nemocných, z toho 96 z kohorty 1 a 365 pacientů z kohorty 2. V týdnu 52 byla klinická remise zjištěna u 60 (39 %) nemocných ze 154 léčených VDZ 1× za 8 týdnů a u 56 nemocných (36,4 %) ze 154 léčených VDZ 1× za 4 týdny. V porovnání s placebem, kde jen 33 osob (21,6 %) ze 153 léčených dosáhlo remise, byl rozdíl vysoce statisticky významný (p < 0,001).
Analýza sekundárních cílů v týdnu 52 ukázala, že většina z nich byla splněna, podíl nemocných s významnou klinickou odpovědí a počet nemocných v remisi bez léčby glukokortikoidy byly v aktivních ramenech s VDZ významně vyšší než u nemocných randomizovaných do placebové větve. Rozdíl v počtu setrvalých klinických remisí nebyl mezi aktivními rameny a placebem prokázán. Jednorázový výskyt protilátek proti VDZ byl zjištěn u 4,1 % probandů a setrvalý výskyt protilátek pouze v 0,4 % případů. Konkomitantní terapie imunosupresivy vedla ke snížení frekvence výskytu protilátek, avšak tato léčba neměla signifikantní vliv na celkový výsledek léčby.
Důležitým zjištěním z této studie bylo, že nemocní, kteří dosáhli odpovědi v 6. týdnu a pokračovali v podávání VDZ v průběhu dalších 46 týdnů, měli počet remisí anebo významných klinických odpovědí v týdnu 52 výrazně vyšší nežli pacienti s klinickou odpovědí na indukční terapii VDZ, kteří pokračovali v udržovací terapii placebem. Negativem v indukční fázi léčby bylo nedosažení statisticky významného rozdílu mezi významnou klinickou odpovědí v týdnu 6 v porovnání s placebem. Polovina z těchto nemocných měla v anamnéze selhání léčby anti‑TNFα v minulosti, z toho přibližně u jedné čtvrtiny pacientů došlo k selhání léčby dvěma nebo více biologickými přípravky.
Mechanismus účinku, který je dán zablokováním vstupu lymfocytů do tkáně, vyžaduje zřetelně více času k dosažení žádaného klinického účinku. Je možné předpokládat, že charakter onemocnění s transmurální distribucí zánětu odpovídá lépe při vyšší expozici léčivu a vyžaduje delší čas léčby. Zkušenosti ze studií s natalizumabem (studie ENACT 1 a 2) potvrdily, že počet dosažených odpovědí a remisí stoupá v průběhu 12 týdnů indukční fáze a dále se zvyšuje i v průběhu udržovací fáze léčby. Je velmi pravděpodobné, že podobná terapeutická „kinetika“ existuje také v případě VDZ. Do 28 týdnů nebyl v udržovací terapii rozdíl mezi aktivními rameny a placebem, a to zřejmě proto, že pozitivní protizánětlivé působení ze dvou indukčních infuzí přetrvávalo relativně dlouhou dobu až do úplné eliminace látky z těla.
Závažné infekce se objevily u 5,5 % nemocných léčených VDZ a u 3,0 % pacientů ze skupiny placeba. Progresivní multifokální leukoencefalopatie se neobjevila u žádného z 1 115 nemocných s Crohnovou nemocí, kteří byli zařazeni do studie a exponováni léčivu. Srovnání dat ze studie GEMINI I a II s výsledky z registru natalizumabu ukázalo vysokou bezpečnost léčby VDZ. U 3 000 nemocných exponovaných léčbě VDZ, z nichž 80 % bylo léčeno imunosupresivy a 30 % bylo léčeno déle než 24 měsíců, nebyl zaznamenán ani jeden případ PML. U léčby natalizumabem představuje incidence PML zhruba jeden případ na 500 léčených, přičemž u pacientů s konkomitantní léčbou imunosupresivy a exponovaných léčbě déle než 24 měsíců je frekvence výskytu PML až jeden případ na 150 léčených [6,7].
Studie GEMINI III
Potřeba odpovědi na některé nejasnosti o účinnosti, dávkování a optimální době k vyhodnocení účinnosti léčby u nemocných s Crohnovou nemocí vedla k vytvoření třetího projektu, který byl zaměřen pouze na indukční fázi léčby – studie GEMINI III. Jednalo se o randomizovanou, dvojitě slepou, placebem kontrolovanou multicentrickou studii fáze III, probíhající v letech 2010 až 2012. Studie byla zaměřena především na pacienty s Crohnovou nemocí, u nichž v minulosti selhala léčba anti‑TNFα. Nemocní byli randomizováni k podávání placeba nebo VDZ v dávce 300 mg i.v. v týdnu 0, 2 a 6. V případě, že v týdnu 10 měli pozitivní léčebnou odpověď, mohli přejít do dlouhodobé, otevřené udržovací fáze studie, ve které byla dávka VDZ podávána jedenkrát měsíčně.
Do studie byli zahrnuti nemocní ve 107 centrech v pěti kontinentech s hodnotou aktivity nemoci vyjádřenou indexem CDAI v rozmezí 220–400. Podobně jako ve studii GEMINI II byly podmínkou zařazení do studie pozitivní endoskopické nebo biologické známky aktivity zánětu. Primárním hodnoceným ukazatelem studie byl poměr nemocných s remisí v týdnu 6 u pacientů se selháním léčby anti‑TNFα. Sekundárním cílovým ukazatelem byl počet nemocných s klinickou remisí v týdnu 6 u všech zařazených nemocných zahrnující také 25 % pacientů, kteří byli k biologické terapii naivní. Dalším sekundárním hodnoceným ukazatelem byl poměr dosažených remisí v týdnu 10 u nemocných, u nichž selhala léčba anti‑TNFα, a u všech zařazených nemocných; podíl nemocných s dosaženou klinickou remisí v týdnu 6 i v týdnu 10, u nichž selhala léčba anti‑TNFα, a u všech randomizovaných nemocných; a podíl dosažené významné klinické odpovědi u pacientů, u nichž selhala léčba anti‑TNFα v týdnu 6 [7,8].
Výsledky studie GEMINI III
Do studie bylo randomizováno celkem 416 nemocných s Crohnovou nemocí, z nichž 315 pacientů prodělalo v minulosti selhání léčby nebo závažné nežádoucí účinky na léčbu anti‑TNFα (76 %), zbývajících 101 nemocných bylo k léčbě anti‑TNFα naivních. Celkem 53 % pacientů užívalo v době randomizace glukokortikoidy, 34 % imunosupresiva a 31 % mesalazin. V populaci nemocných, kteří již byli v minulosti léčeni přípravky anti‑TNFα, bylo 66 % těch, jimž byly podávány dva a více přípravků biologické léčby.
Primární cíl (počet dosažených klinických remisí v týdnu 6) nebyl splněn. V aktivním rameni se nacházelo v remisi 15,2 % nemocných, v placebové větvi 12,1 % hodnocených nemocných, rozdíl nebyl statisticky významný (p = 0,433). Při srovnání všech pacientů (včetně těch, kterým nebyla biologická léčba nikdy podávána) byl rozdíl v 6. týdnu mezi placebem a aktivním ramenem statisticky významný, 12,1 % pacientů ve skupině placeba vs. 19,1 % ve skupině s léčbou VDZ (p = 0,048). Pochopitelně nejlepších výsledků bylo docíleno u nemocných, kteří byli k biologické léčbě naivní; z nich dosáhlo 12 % remise při podávání placeba, zatímco v rameni s aktivní léčbou byla remise zjištěna u 31,4 % pacientů.
Výsledky v týdnu 10 však vykazovaly oproti týdnu 6 nápadné odlišnosti. Remise u nemocných se selháním léčby anti‑TNFα byla v desátém týdnu zjištěna u 26,6 % pacientů v aktivním rameni, ale jen u 12,1 % pacientů ze skupiny s placebem (p = 0,001). Stejně výrazné rozdíly byly ve skupině nemocných naivních k léčbě anti‑TNFα; 35,3 % pacientů ve skupině s aktivní léčbou v porovnání s 16,0 % pacientů ve skupině s placebem (p < 0,001). Srovnání současně dosažené remise v obou týdnech 6 a 10 u všech pacientů prokázalo významné rozdíly mezi aktivní léčbou a placebem (15,3 % vs. 8,2 %; p = 0,025); ve skupině pacientů, u nichž selhala léčba anti‑TNFα, však nebyl zjištěný rozdíl signifikantní (12,0 % vs. 8,3 %; p = 0,276). U nemocných naivních k biologické léčbě byl mezi ramenem s aktivní léčbou a skupinou, v níž bylo podáváno placebo, potvrzen významný rozdíl (25,5 % vs. 8,0 %). Významná klinická odpověď v týdnu 6 byla splněna ve všech ohledech, tj. u všech nemocných (39,2 % vs. 22,7 %; p = 0,002), ve skupině pacientů, u nichž selhala léčba anti‑TNFα (39,2 % vs. 22,3 %; p = 0,001), a numericky byl vyšší rozdíl i ve skupině naivních pacientů (39,2 % vs. 24,0 %; p = 0,088) [8].
Provedená studie GEMINI III ukázala, že nemocní s předcházejícím selháním léčby anti‑TNFα dosahují významné klinické odpovědi na terapii VDZ až mezi týdnem 6 a 10. Téměř dvojnásobný nárůst podílu pacientů dosahujících klinické remise mezi týdnem 6 a 10 (15,2 % vs. 26,6 %) je toho jasným dokladem. Důležité je podotknout, že poměr nemocných s dosaženou remisí při podávání placeba byl v týdnu 6 a 10 totožný. Z hlediska všech nemocných, kteří byli do studie randomizováni, se potvrdilo, že celkové výsledky jsou velmi příznivé a dokládají statisticky významný rozdíl mezi aktivní léčbou a placebem. Tyto pozitivní výsledky byly dány vysokou mírou odpovědi v šestém týdnu u nemocných, kteří byli k biologické léčbě naivní. V podskupině nemocných naivních k biologické léčbě byl zaznamenán významně nižší počet operací, kratší trvání choroby a lze u těchto nemocných předpokládat nižší výskyt strukturálních a ireverzibilních změn na střevě v porovnání s pacienty, u nichž v minulosti došlo k selhání biologické léčby.
Předpokládané postavení vedolizumabu v klinické praxi
Výsledky studie GEMINI I potvrdily, že indukční terapie VDZ je účinnou léčebnou modalitou u nemocných s ulcerózní kolitidou, u nichž došlo k selhání nejen konvenční terapie glukokortikoidy, imunosupresivy, ale také k selhání biologické léčby anti‑TNFα. Z hlediska krátkodobé klinické účinnosti bylo v šestém týdnu dosaženo nižšího podílu pozitivních odpovědí a remisí, než tomu bylo u biologicky naivních pacientů ve studiích ACT‑1 a ACT‑2 při podávání infliximabu. V osmém týdnu po třech infuzích infliximabu v dávce 5 mg/kg bylo dosaženo klinické odpovědi u 61,4 % a u 38,8 % pacientů. V porovnání se subkutánně podávanými přípravky, jejichž účinek byl také hodnocen v týdnu 6 – adalimumab (studie ULTRA‑1 a ULTRA‑2) a golimumab (studie PERSUIT SC) – byly výsledky srovnatelné. Dominující jsou však výsledky podávání VDZ v dlouhodobé udržovací terapii. Počet remisí v 52. týdnu, který dosáhl 44,8 %, respektive 41,4 %, nápadně převyšuje výsledky studie ACT‑1 (25,6 %), ULTRA‑2 (17,3 %) a PURSUIT SC (27,8 %) [7]. Výsledky studií se subkutánně podávanými monoklonálními protilátkami anti‑TNFα u pacientů s ulcerózní kolitidou vykázaly signifikantní účinnost proti placebu v indukční i udržovací fázi klinického hodnocení, ale z praktického hlediska byly výsledky daleko méně přesvědčivé, než tomu bylo v případě infliximabu.
Nejzásadnější účinek nové terapie s VDZ je spatřován právě v dlouhodobé udržovací terapii, jejíž dosavadní výsledky jsou ze všech biologických léčiv nejpřesvědčivější, i když srovnání jednotlivých studií a souborů nemocných je velmi obtížné. Je zde nápadný rozdíl v „kinetice“ terapeutické odpovědi oproti léčbě anti‑TNFα, jejíž protizánětlivý účinek (zvláště v případě infliximabu) je nápadný hned po první infuzi a kulminuje mezi 10. až 14. týdnem od zahájení léčby. Bohužel, při další léčbě dochází ke snižování intenzity odpovědi a často se objevuje druhotná ztráta odpovědi. Největší počet nemocných přestává odpovídat na terapii infliximabem v prvním roce léčby (cca 20–30 %) a v dalších letech konstantně dochází ke ztrátě 10–15 % původně příznivě odpovídajících pacientů. V případě VDZ se ukázalo, že s délkou podávání klinický účinek VDZ spíše narůstá.
Druhou velkou výhodou podávání VDZ je, že k dosažení optimální účinnosti není důležitá imunosupresivní léčba, která v případě infliximabu potlačením produkce protilátek proti léčivu snižuje clearance léčiva a zvyšuje jeho terapeutickou účinnost. Nevýhody kombinované terapie jsou zjevné a spočívají v silnějším celkovém imunosupresivním účinku a ve vyšším riziku oportunních infekcí.
Nevýhodou terapie VDZ oproti léčbě anti‑TNFα je pomalejší nástup účinku, což znevýhodňuje podání léčiva u nemocných s vysokou zánětlivou aktivitou, dále u pacientů, kde je třeba odvrátit vznik komplikace nebo nutnost chirurgické intervence. V tomto směru pravděpodobně VDZ nenahradí dříve využívaný ciclosporin A anebo v pozdějších letech častěji aplikovaný infliximab. Jinou terapeutickou nevýhodou může být vysoká selektivita VDZ k gastrointestinálnímu traktu a pravděpodobně jen malá účinnost u nemocných s mimostřevními projevy nemoci, jako je iridocyklitida, pyoderma gangrenosum nebo enteropatická artritida I. typu.
Vzhledem k minimálním klinickým zkušenostem nemáme zatím žádná data týkající se bezpečnosti léčby VDZ u těhotných žen a také u dětských pacientů. Podle všeho by vhodnými pacienty k zahájení terapie VDZ mohli být: a) nemocní s vleklou aktivitou ulcerózní kolitidy, u kterých se nepodařilo docílit remise při léčbě glukokortikoidy a imunosupresivy; b) pacienti, kteří ztratili odpověď nebo nereagují na podávání léčby anti‑TNFα; c) pacienti s výraznými nežádoucími účinky léčby azathioprinem (pankreatitida, myelosuprese) nebo přípravky anti‑TNFα (recidivující oportunní infekce). V tomto ohledu se zdá být pozice VDZ velmi silná a minimálně srovnatelná s ostatními biologickými léčivy ze skupiny protilátek anti‑TNFα.
U nemocných s Crohnovou nemocí byla účinnost VDZ méně impresivní, což může ukazovat na odlišný patofyziologický mechanismus zánětu a jeho transmurální charakter. V indukční fázi by měly být užity tři dávky léčiva v týdnu 0, 2 a 6 a klinická odpověď by měla být hodnocena až v týdnu 10. Podle některých autorů je možné dokonce použít v rámci indukce ještě čtvrtou infuzi v 10. týdnu a odpověď hodnotit až v týdnu 14, a to především u nemocných, kteří nereagují nebo přestali odpovídat na léčbu přípravky anti‑TNFα.
Jinými slovy, u pacientů s agresivními formami Crohnovy nemoci je v porovnání s pacienty s ulcerózní kolitidou potřeba větší expozice léčivu a delší doba k dosažení požadovaného cíle. Výsledky studie GEMINI III představují silný argument pro využití VDZ jako účinné biologické terapie druhé linie u pacientů s komplikovaným průběhem Crohnovy nemoci.
Zjevnou, byť asi jen dočasnou nevýhodou VDZ jsou chybějící data o účinnosti v léčení perianální nemoci. Protože význam aktivovaných α4β7‑pozitivních lymfocytů je v patogenezi této formy Crohnovy nemoci jen okrajový, je dosti pravděpodobné, že právě v této indikaci zůstanou přípravky anti‑TNFα stále nejúčinnější léčebnou modalitou.
Projekt GEMINI ukázal, že terapeutickou odpověď na léčbu VDZ budeme muset opřít o jiné, převážně klinické parametry, protože monitorování tzv. biologických markerů aktivity onemocnění v tomto ohledu pokulhává. Vliv na snížení sérové koncentrace CRP byl ve studiích s VDZ vyjádřen jen mírně, zvláště v indukční fázi léčby; na rozdíl od protilátek anti‑TNFα, které mají přímý vliv na jaterní buňky, kde snižují syntézu CRP zablokováním membránově vázaného TNFα a také přímým vlivem na produkci CRP v tukové tkáni a v mezenteriu. Snížení tvorby CRP při aplikaci VDZ pravděpodobně představuje až druhotný výsledek léčby, až po významném snížení zánětlivé aktivity v důsledku inhibice migrace lymfocytů. Jak bylo pozorováno ve studii GEMINI II, je snižování hodnot CRP velmi pomalé a postupné a trvá po celou dobu podávání udržovací fáze léčby a nelze podle změny hodnot CRP na konci indukční fáze léčby spolehlivě predikovat její klinický účinek.
Zavedení VDZ do léčby Crohnovy nemoci a ulcerózní kolitidy je vítaným a dlouho očekávaným krokem, který nepochybně přispěje k posílení našich terapeutických možností a ke zvýšení celkové účinnosti léčby u pacientů s IBD. Z výše uvedených klinických studií lze jen rámcově odhadovat, jaké bude postavení VDZ v klinické praxi. To podstatné přinese až dlouhodobá klinická zkušenost, která také určí optimální typ pacienta, jemuž přinese léčba druhou generací antiintegrinových protilátek dlouhodobý prospěch. Nemalý vliv na postavení léku v léčebném armamentariu bude mít také dostupnost léčiva, která bude určena především ekonomickou nákladností léčby [9].
Seznam použité literatury
- [1] Bortlík M. Vedolizumab nová anti integrinová protilátka s vysokou gastrointestinální selektivitou. Gastroent Hepatol 2014; 68: 481–484.
- [2] Raine T. Vedolizumab for inflammatory bowel disease: Changing the game, or more of the same? UEG Journal 2014; 5: 333–344.
- [3] Sanborn WJ, Colombel JF, Enns R, et al. Natalizumab induction and maintenance therapy for Crohn´s disease. N Engl J Med 2005; 353: 1912–1925.
- [4] Feagan BC, Rutgeerts P, Sands BE, et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med 2013; 369: 699–710.
- [5] Lukáš M. Vedolizumab v léčbě ulcerozní kolitidy. Gastro Hepatol 2015; 69: 29–32.
- [6] Sandborn WJ, Feagan BC, Rutgeerts P, et al. Vedolizumab for induction and maintenance therapy for Crohn´s disease. N Engl J Med 2013; 369: 711–721.
- [7] Lukáš M. Vedolizumab v léčbě Crohnovy nemoci. Gastro Hepatol 2015; 69: v tisku.
- [8] Sands BE, Feagan BG, Rutgeerts P, et al. Effects of vedolizumab induction therapy for patients with Crohn´s disease in whom tumor necrosis factor antagonist treatment failed. Gastro 2014; 147: 618–627.
- [9] Lukáš M. Biologická léčba idiopatických střevních zánětů. In: Biologická léčba zánětlivých autoimunitních onemocnění v revmatologii, gastroenterologii a dermatologii. Edited by: Karel Pavelka, Petr Arenberger, Milan Lukáš, Tomáš Zima, Tomáš Doležal a Marta Olejárová. Praha: Grada, 2014.