Zaostřeno Castlemanova choroba
OBSAH
 Castlemanova choroba - editorial
Castlemanova choroba - editorial
Castlemanova choroba – skrytá hrozba mezi námi
Castlemanova choroba není benigní onemocnění
Od vstupu siltuximabu jsme neztratili jediného pacienta - rozhovor
Idiopatická Castlemanova choroba očima pacientky
Kazuistika pacienta s Castlemanovým syndromem z Klinického centra Univerzity v Debrecínu
Castlemanova choroba
Castlemanova choroba je vzácné onemocnění. Orphanet uvádí již přes 11 000 kódů vzácných chorob. V reálné medicíně počítáme odhadem se sedmi tisíci diagnózami. I v České republice žije kolem 500 tisíc lidí se vzácným onemocněním. A netroufnu si odhadnout, kolik je nediagnostikovaných pacientů. To je problém i lymfoproliferativního onemocnění s nálezem benigního histologického obrazu, ale mnohdy s maligním průběhem – Castlemanovy choroby. Ze zkušeností pacientů i z obecných pravidel vyplývá, že na vzácnou chorobu se musí upozornit, a to zejména existuje‑li pro ni léčba, aby byla včas diagnostikována a léčena. Žádný odborník totiž nemůže znát a diagnostikovat každou jednu ze sedmi tisíc vzácných chorob. Proto publikujeme toto speciální vydání Zaostřeno – Castlemanova choroba. Přinášíme v něm soubor článků, které představí toto onemocnění z různých úhlů pohledu.
Průkopník na poli Castlemanovy choroby – doktor David Fajgenbaum – sám na idiopatickou multicentrickou chorobu málem pětkrát zemřel. Nyní (doufám, že stále) je v dlouhodobé remisi, oženil se a má dvě děti.
O svém boji s nemocí i o urputné snaze zajistit nejen sobě, ale všem pacientům správnou léčbu píše ve své knize Chasing My Cure: A Doctor᾿s Race to Turn Hope Into Action (Hon za léčbou: doktorův závod o proměnu naděje v činy) [1].
V úvodu (kromě jiného) píše: „Když jsem se stal lékařem, byl jsem svědkem nevyléčitelné nemoci a neutišitelného smutku – moje matka zemřela na rakovinu mozku. Od vysoké školy jsem však stále optimistou, pokud jde o sílu vědy a medicíny najít odpovědi a léčbu (…) Během posledních let jsem měl spoustu času na to, abych o sobě přemýšlel. Jedna věc, kterou jsem se naučil, je, že každý z nás, kdo si obléká bílý plášť, má úzký vztah k pojmu autorita. Všichni se snažíme být důvěryhodnými v zodpovídání naléhavých otázek… Ani jeden z nás neví všechno, co je k vědění. Ani zdaleka ne. Čas od času můžeme podávat mistrovské výkony – a jen málo vybraných může být skutečně mistry v určitých specializacích – ale z velké části přijímáme své limity… Pravda je, že nikdo neví všechno, ale to není ten problém. Problém je, že o některých věcech nikdo nic neví, nic se nedělá, aby se to změnilo, a upřímně, někdy se medicína může mýlit. Moje dobrodružství jako lékaře i pacienta mě naučila mnohé o často nespravedlivém nesouladu mezi tím nejlepším, co může věda nabídnout, a naší křehkou dlouhověkostí, mezi myšlenkami a modlitbami a zdravím a blahobytem. Tohle je příběh o tom, jak jsem zjistil, že Ježíškovi zástupci v medicíně neexistují, nepracují na mém dárku a nedodají mi lék. Je to také příběh o tom, jak jsem pochopil, že naděje nemůže být pasivní pojem. Je to volba a síla; doufat v něco vyžaduje víc než vyslat přání do vesmíru a čekat, až se to stane. Naděje by měla inspirovat k akci. A když inspiruje k akci v medicíně a vědě, může se tato naděje stát realitou, která přesahuje vaše nejdivočejší sny. V podstatě je to příběh o umírání, ze kterého, doufám, se můžete naučit žít.“
Úryvky z knihy Chasing My Cure vybrala
MUDr. Marta Šimůnková
David Fajgenbaum, MD, MBA, MSc., (Pennsylvania, Center for Cytokine Storm Treatment & Laboratory; Associate Director, Orphan Disease Center, University of Pennsylvania; Co‑Founder & President, Castleman Disease Collaborative Network [CDCN]; Co‑Founder, Every Cure) přijal na základě vlastní zkušenosti za svůj životní program a cíl věnovat se výzkumu, publikacím i publicitě, pacientské advokacii, vývoji nových léčebných metod v oblasti idiopatické multicentrické Castlemanovy choroby.
[1] Fajgenbaum DC. Chasing My Cure. Random House USA Inc. 2021.
Castlemanova choroba – skrytá hrozba mezi námi
MUDr. Dominika Écsiová
1. interní hematologická klinika FN a LF UK Hradec Králové
Úvod
Castlemanova nemoc je vzácné a závažné onemocnění lymfatického systému. Vyznačuje se nadměrným zvětšením lymfatických uzlin, laboratorními abnormalitami a může vést k těžkým komplikacím. Navzdory své závažnosti je často diagnostikována nesprávně nebo pozdě kvůli překrývání příznaků s jinými onkologickými a autoimunitními onemocněními. Tento článek poskytuje podrobný přehled epidemiologie, patofyziologie a současných léčebných strategií idiopatické multicentrické Castlemanovy choroby (iMCD), včetně inovativních cílených terapií.
Obecný rámec problematiky
Poprvé byla Castlemanova nemoc popsána Benjaminem Castlemanem v roce 1954, multicentrická forma byla identifikována až v roce 1978 [1].
Castlemanova nemoc představuje spektrum neklonálních lymfoproliferativních onemocnění se společnou histopatologií. Jedná se o vzácné a často zničující onemocnění, které může způsobit dysfunkci více orgánů. Multicentrická forma (MCD) je obzvláště prognosticky nepříznivá, necelá polovina léčených pacientů nemá dostatečnou léčebnou odpověď na první linii terapie a třetina pacientů s idiopatickou formou nemoci umírá do pěti let od diagnózy [2].
Patogeneze onemocnění není zcela objasněna, existují pouze teorie jejího vzniku. Jsou známy tři hlavní hypotézy patogeneze, které vysvětlují různé možné mechanismy vedoucí k rozvoji tohoto onemocnění.
První je virová hypotéza: Tato teorie předpokládá, že přítomnost specifických virů způsobuje hypercytokinemii prostřednictvím signalizace interleukinu 6 (IL‑6). IL‑6 je prozánětlivý cytokin, který hraje klíčovou roli v imunitní odpovědi a jeho nadprodukce může vést k nekontrolovanému růstu lymfatických uzlin a zánětlivým reakcím [1].
Další hypotézou je hypotéza paraneoplastického syndromu: Podle této hypotézy jsou prozánětlivé cytokiny uvolňovány v důsledku přítomnosti benigního nebo maligního nádoru. Nádory mohou indukovat produkci těchto cytokinů, což vede k systémovým zánětlivým odpovědím a patologickým změnám v lymfatické tkáni [3].
Poslední zvažovanou je autoimunitní hypotéza: Třetí teorie navrhuje, že hypercytokinemie je způsobena antigen prezentujícími buňkami a autoprotilátkami. Tento mechanismus zahrnuje abnormální aktivaci imunitního systému, kde vlastní imunitní buňky těla napadají zdravé tkáně, což vede k chronickému zánětu a proliferaci lymfatických buněk [3].
Každá z těchto hypotéz přispívá k našemu porozumění komplexní patogenezi Castlemanovy nemoci a může mít významné dopady na diagnostiku a léčbu tohoto onemocnění.
Klinické spektrum Castlemanovy nemoci
Castlemanova nemoc se dělí na dvě hlavní formy – unicentrickou a multicentrickou –, přičemž každá má své specifické klinické a patologické charakteristiky.
Unicentrická forma (UCD) postihuje pouze jednu lymfatickou uzlinu nebo region a je charakterizována lokalizovanou lymfadenopatií. Velmi často je asymptomatická a může být náhodně objevena během vyšetření z jiných důvodů. Typicky chybějí systémové příznaky, které jsou přítomny u multicentrické formy. Při sledování 404 pacientů s unicentrickou formou byla pozorována jako nejčastější místa postižení mediastinum (29 %), krk (23 %), břicho (21 %) a retroperitoneum (17 %) [4]. Chirurgická resekce postižené uzliny je obvykle kurativní. U 10–13 % pacientů je lymfadenopatie neresekovatelná nebo po excizi dojde k recidivě a měla by být v takovém případě léčena jako multicentrická forma nemoci [5]. Při absenci jiných známek onemocnění může být zvolena strategie „watch and wait” nebo jiné intervence, jako je radioterapie.
Multicentrická forma (MCD) zahrnuje více lymfatických uzlin v různých oblastech těla a je spojena s výraznými systémovými příznaky, jako jsou horečka, noční pocení, úbytek hmotnosti a únava. MCD může být dále rozdělena na několik podtypů.
HHV‑8 asociovaná MCD: Tato forma je spojena s infekcí virem lidského herpesviru 8 (HHV‑8) a často postihuje osoby s oslabeným imunitním systémem, jako jsou pacienti s HIV. Léčba zahrnuje antiretrovirovou terapii a antivirotika [6].
MCD spojená s POEMS syndromem: POEMS je zkratka pro polyneuropatii, organomegalii, endokrinopatii, monoklonální gamapatii a kožní změny. Tento syndrom je asociován s multicentrickou Castlemanovou nemocí a vyžaduje komplexní přístup k léčbě [7].
Idiopatická MCD (iMCD): Tento podtyp nemá známou virovou etiologii a může být dále rozdělen na subtyp TAFRO (trombocytopenie, anasarka, febrilie, retikulinová fibróza a organomegalie) a iMCD NOS (nespecifikovaný podtyp) [8,9]. Idiopatická MCD je závažná forma s různorodými klinickými projevy a vyžaduje specifickou léčbu, která může zahrnovat imunoterapii a inhibitory IL‑6 [10].
Toto rozdělení Castlemanovy nemoci umožňuje přesnější diagnostiku a léčbu na základě specifických charakteristik každého typu, což může vést ke zlepšení klinických výsledků a prognózy pacientů.
Diagnostika multicentrické Castlemanovy nemoci
Multicentrická Castlemanova choroba postihuje více skupin lymfatických uzlin, nejčastěji v oblasti krku, mediastina, axil a retroperitonea. Častěji bývá provázena rozmanitými symptomy, včetně krvácivých příznaků při trombocytopenii, anemického syndromu při anémii a B symptomů (febrilie, noční pocení, hmotnostní úbytek). Idiopatická multicentrická Castlemanova choroba tvoří více než 50 % případů MCD a není asociována se známou virovou infekcí HIV nebo HHV‑8. Pacienti s touto formou onemocnění vykazují odlišné léčebné odpovědi ve srovnání s non‑idiopatickou MCD [11].
Non‑idiopatická MCD je často sekundární k infekci HIV nebo HHV‑8 a může být součástí POEMS syndromu. Akronymu POEMS odpovídá výskyt specifických příznaků, jako jsou polyneuropatie, organomegalie, endokrinopatie, monoklonální gamapatie a kožní změny. V rámci Castlemanovy choroby se jedná o zvláště závažnou manifestaci, která může komplikovat průběh onemocnění. Diagnostika POEMS syndromu u Castlemanovy nemoci vyžaduje pečlivé hodnocení typických příznaků a potvrzení etiopatogeneze onemocnění, což může být komplikováno častým úvodním těžkým rozpoznáváním těchto příznaků a zaměňováním za jiné neurologické poruchy nebo jiná interní onemocnění. Důležité je zaměření na typické znaky a použití sofistikovaných diagnostických metod, včetně biopsie a zobrazovacích vyšetření, pro potvrzení diagnózy. Léčba zahrnuje radioterapii, chemoterapii zaměřenou na plazmocelulární dyskrazie a symptomatický management periferní neuropatie [12].
Pro diagnózu a hodnocení Castlemanovy nemoci se doporučuje řada vyšetření, která zahrnují jak základní, tak doplňkové testy, jež jsou užitečné za určitých okolností.
Základní vyšetření:
- Fyzikální vyšetření: Zvláštní pozornost je věnována oblastem lymfatických uzlin, včetně uzlin Waldeyerova okruhu, a velikosti jater a sleziny.
- Anamnéza se zaměřením na přítomnost B příznaků, hodnocení celkového zdravotního stavu a schopnosti provádět běžné denní činnosti.
- Krevní obraz s mikroskopickým diferenciálním rozpočtem bílých krvinek k vyloučení jiné malignity, která může mít podobné příznaky.
- Komplexní metabolický panel: Testy na hodnocení metabolických funkcí, včetně hladiny elektrolytů, glukózy a funkce ledvin.
- LDH, C‑reaktivní protein, markery nádorového rozpadu.
- Beta‑2‑mikroglobulin, elektroforéza sérových proteinů a elektroforéza moči s imunofixací, sérové lehké řetězce, kvantitativní imunoglobuliny: Testy na hodnocení bílkovin a imunitních funkcí.
- Testování na HIV, HHV‑8, hepatitidu B, hepatitidu C, EBV a CMV: Virologické testy pro zjištění možných infekcí.
- PET/CT vyšetření (preferováno) nebo CT hrudníku, břicha a pánve s kontrastem: Zobrazovací techniky pro diagnostiku a hodnocení rozsahu onemocnění a také k určení vhodné uzliny k diagnostické exstirpaci.
- Těhotenský test u pacientek v plodném věku: Pokud je plánována chemoterapie nebo radioterapie.
Užitečné za určitých okolností:
- Biopsie kostní dřeně a aspirát: Zvláště u pacientů s TAFRO syndromem pro hodnocení retikulinové fibrózy kostní dřeně.
- Echokardiogram nebo MUGA sken: Pokud je indikována léčba založená na antracyklinech.
- Testování na IgG4, sIL‑6, sIL‑10, VEGF, kyselinu močovou, feritin: Doplňkové testy na zánětlivé a růstové faktory. V mnoha laboratořích nemusejí být dostupné, nicméně pro diagnostiku nejsou klíčové.
- Diskuse o zachování plodnosti: Konzultace týkající se možností zachování plodnosti před zahájením léčby.
Tato vyšetření představují pouze jednu část celkového diagnostického procesu. Zásadní význam má přítomnost specifických histologických změn, které společně s klinickým obrazem a laboratorními výsledky tvoří základ pro přesnou diagnostiku a efektivní řízení léčby Castlemanovy nemoci [13].
Studie uvádí, že diferenciální diagnostika Castlemanovy nemoci je náročná kvůli překrývání klinických projevů s jinými onemocněními, jako jsou infekce, autoimunitní choroby a maligní nádory. To vede k tomu, že mnoho případů je původně mylně diagnostikováno jako jiné nemoci. Přesný podíl chybných diagnóz se může lišit v závislosti na typu nemoci [14].
Léčba MCD asociované s HIV nebo HHV‑8
Pro léčbu multicentrické Castlemanovy nemoci spojené s HIV nebo HHV‑8 se doporučuje komplexní přístup zahrnující imunoterapii a antiretrovirovou terapii. Důležité je, že u pacientů s HIV je prioritní zahájení kombinované antiretrovirové terapie. Pro samotné léčení MCD je obvyklé použití rituximabu, což je monoklonální protilátka proti CD20. Může být podáván samostatně nebo v kombinaci s lipozomálním doxorubicinem nebo prednisonem. Tento režim je zvláště účinný vzhledem ke své schopnosti adresovat patologické B buňky, které jsou běžně přítomny u této formy MCD. U pacientů, kteří jsou pozitivní na HHV‑8, ale nemají HIV, může být využit podobný léčebný režim. Rituximab je často kombinován s chemoterapií pro zvýšení účinnosti léčby, což může zahrnovat režimy jako CHOP (cyklofosfamid, doxorubicin, vinkristin a prednison) pro řešení agresivnějších forem onemocnění [15].
Léčba idiopatické MCD
Idiopatická MCD je forma nemoci bez známé infekční etiologie, jako je HHV‑8 nebo HIV a vyžaduje odlišný přístup. U těchto pacientů je často používán siltuximab, monoklonální protilátka proti IL‑6, který hraje klíčovou roli v patogenezi nemoci tím, že stimuluje zánětlivou reakci a růst lymfoidních buněk. Siltuximab se ukázal být účinný ve stabilizaci nemoci a zmírnění příznaků [16,17]. Další léčebné možnosti zahrnují rituximab, thalidomid, cyklofosfamid a prednison a mohou být použity samostatně nebo v kombinaci v závislosti na závažnosti onemocnění a léčebné odpovědi [18].
Donedávna neexistovala žádná diagnostická kritéria ani léčebné možnosti. Idiopatická MCD je vzácné onemocnění s roční incidencí ve Spojených státech amerických < 1 000 pacientů [19]. V České republice není přesný počet znám, můžeme se o něm pouze dohadovat. Nedostatek údajů z reálné praxe ztěžuje přístup k informacím o léčebných odpovědích u pacientů mimo klinické studie. Diagnostika je obtížná pro překrývající se symptomy s jinými malignitami, autoimunitními nemocemi a infekcemi. Zásadní je správný diagnostický postup, včetně excize celé lymfatické uzliny pro histopatologické vyšetření a komplexní klinické a laboratorní vyšetření pacienta [13].
Časté klinické příznaky:
Systematický přehled, který identifikoval 128 pacientů s potvrzenou idiopatickou multicentrickou Castlemanovou nemocí, analyzoval běžné klinické příznaky a sestavil výčet častých projevů:
- Generalizovanou lymfadenopatii mělo 100 % pacientů.
- Flu‑like syndrom – febrilie se vyskytly u 26–52 % pacientů.
- Noční pocení bylo pozorováno u 10–62 % pacientů.
- Nezamýšlený úbytek hmotnosti udávalo 16–72 % pacientů.
- Edém, ascites a anasarka se vyskytly u 23–78 % pacientů.
- Hepatomegalie a/nebo splenomegalie byly diagnostikovány u 33–78 % pacientů [2].
Život ohrožující příznaky:
- Anasarka a pancytopenie.
- Neurologické příznaky: kóma, křeče, cévní mozkové příhody.
- Multiorgánové selhání.
- Nedávná analýza 31 pacientů ukázala progresi u všech neléčených pacientů (8 z 8) [8].
U idiopatické formy nemoci existuje trojnásobně zvýšené riziko přítomnosti malignity, především lymfomu [3].
Léčebný algoritmus pro idiopatickou multicentrickou Castlemanovu nemoc shrnuje podrobně následující text MUDr. Šimůnkové, stejně tak výsledky studií s cílenou a necílenou léčbou tohoto onemocnění [20–25].
Závěr
Castlemanova nemoc je často obtížně diagnostikovatelná kvůli své heterogenitě a překrývání příznaků s jinými nemocemi, jako jsou infekce, autoimunitní onemocnění a maligní nádory. Tento fakt vede k tomu, že mnoho případů je v úvodu mylně diagnostikováno. Diferenciální diagnostika je náročná a vyžaduje komplexní přístup zahrnující histopatologii, laboratorní testy a zobrazovací techniky. Tyto kroky jsou klíčové pro správné určení diagnózy a efektivní léčbu, což může výrazně zlepšit prognózu pacientů. Dodržení diagnostických postupů a zvýšení povědomí o Castlemanově nemoci mezi lékaři jsou nezbytné k minimalizaci rizika chybných diagnóz a k zajištění včasné a správné léčby.
Literatura
[1] Dispenzieri A, Fajgenbaum DC. Overview of Castleman disease. Blood 2020; 135: 1353–1364.
[2] Liu AY, Nabel CS, Finkelman BS, et al. Idiopathic multicentric Castlemanʼs disease: a systematic literature review. Lancet Haematol 2016; 3: e163–175.
[3] Fajgenbaum DC, van Rhee F, Nabel CS. HHV‑8‑negative, idiopathic multicentric Castleman disease: novel insights into biology, pathogenesis, and therapy. Blood 2014; 123: 2924–2933.
[4] Talat N, Belgaumkar AP, Schulte KM. Surgery in Castlemanʼs disease: a systematic review of 404 published cases. Ann Surg 2012; 255: 677–684.
[5] Mohan M, Meek JC, Meek ME, et al. Combinatorial treatment for unresectable unicentric Castleman disease. Eur J Haematol 2021; 107: 484–488.
[6] Barlingay G, Findakly D, Hartmann C, Amar S. The Potential Clinical Benefit of Tocilizumab Therapy for Patients with HHV‑8‑infected AIDS‑related Multicentric Castleman Disease: A Case Report and Literature Review. Cureus 2020; 12: e7589.
[7] Sobas MA, Alonso Vence N, Diaz Arias J, et al. Efficacy of bortezomib in refractory form of multicentric Castleman disease associated to poems syndrome (MCD‑POEMS variant). Ann Hematol 2010; 89: 217–219.
[8] Iwaki N, Fajgenbaum DC, Nabel CS, et al. Clinicopathologic analysis of TAFRO syndrome demonstrates a distinct subtype of HHV‑8‑negative multicentric Castleman disease. Am J Hematol 2016; 91: 220–226.
[9] Nishimura Y, Hanayama Y, Fujii N, et al. Comparison of the clinical characteristics of TAFRO syndrome and idiopathic multicentric Castleman disease in general internal medicine: a 6‑year retrospective study. Intern Med J 2020; 50: 184–191.
[10] van Rhee F, Casper C, Voorhees PM, et al. Long‑term safety of siltuximab in patients with idiopathic multicentric Castleman disease: a prespecified, open‑label, extension analysis of two trials. Lancet Haematol 2020; 7: e209–e217.
[11] Fajgenbaum DC, Uldrick TS, Bagg A, et al. International, evidence‑based consensus diagnostic criteria for HHV‑8‑negative/idiopathic multicentric Castleman disease. Blood 2017; 129: 1646–1657.
[12] Dispenzieri A. POEMS Syndrome: 2019 Update on diagnosis, risk‑stratification, and management. Am J Hematol 2019; 94: 812–827.
[13] National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: Castleman Disease [Internet]. Version 1.2024. Dostupné na: https://www.nccn.org/professionals/physician_gls/pdf/castleman.pdf
[14] Hu S, Li Z, Wang H, et al. Clinical features and treatment outcomes of Castleman disease in children: a retrospective cohort in China. Eur J Pediatr 2023; 182: 5519–5530.
[15] National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology: HIV‑Related Cancer [Internet]. Version 1.2023. Dostupné na: https://www.nccn.org/professionals/physician_gls/pdf/hiv.pdf
[16] Yoshizaki K, Murayama S, Ito H, Koga T. The Role of Interleukin‑6 in Castleman Disease. Hematol Oncol Clin North Am 2018; 32: 23–36.
[17] Tanaka T, Kishimoto T. The biology and medical implications of interleukin‑6. Cancer Immunol Res 2014; 2: 288–294.
[18] Dong Y, Zhang L, Nong L, et al. Effectiveness of rituximab‑containing treatment regimens in idiopathic multicentric Castleman disease. Ann Hematol 2018; 97: 1641–1647.
[19] van Rhee F, Oksenhendler E, Srkalovic G, et al. International evidence‑based consensus diagnostic and treatment guidelines for unicentric Castleman disease. Blood Adv 2020; 4: 6039–6050.
[20] van Rhee F, Rosenthal A, Kanhai K, et al. Siltuximab is associated with improved progression‑free survival in idiopathic multicentric Castleman disease. Blood Adv 2023; 7: 1604–1605.
[21] Kawabata H, Kotani S, Matsumura Y, et al. Successful treatment of a patient with multicentric Castlemanʼs disease who presented with thrombocytopenia, ascites, renal failure and myelofibrosis using tocilizumab, an anti‑interleukin‑6 receptor antibody. Intern Med 2013; 52: 1503–1507.
[22] Ramasamy K, Gandhi S, Tenant‑Flowers M, et al. Rituximab and thalidomide combination therapy for Castleman disease. Br J Haematol 2012; 158: 421–423.
[23] Dispenzieri A, Armitage JO, Loe MJ, et al. The clinical spectrum of Castlemanʼs disease. Am J Hematol 2012; 87: 997–1002.
[24] Sitenga J, Aird G, Ahmed A, Silberstein PT. Impact of siltuximab on patient‑related outcomes in multicentric Castlemanʼs disease. Patient Relat Outcome Meas 2018; 9: 35–41.
[25] Brandstadter JD, Fajgenbaum DC. How we manage idiopathic multicentric Castleman disease. Clin Adv Hematol Oncol 2022; 20: 564–571.
Castlemanova choroba není benigní onemocnění
S Castlemanovou chorobou se mohou setkat lékaři všech specializací, kteří řeší příčinu tzv. uzlinového syndromu. Unicentrická forma tohoto progresivního lymfoproliferativního onemocnění postihuje jednu lymfatickou uzlinu nebo skupinu uzlin, multicentrická více skupin uzlin. Závažnost idiopatické multicentrické Castlemanovy choroby se obecně podceňuje, ale jak ukazují mortalitní data, jde o vážné a bez léčby progredující až fatální onemocnění.
Castlemanova choroba je progresivní lymfoproliferativní onemocnění. Unicentrická forma (UCD) postihuje jednu lymfatickou uzlinu nebo jednu skupinu uzlin nejčastěji v oblasti břicha a krku a obvykle se léčí chirurgickou excizí [1–3]. U 10–13 % pacientů je UCD neresekovatelná nebo po excizi dojde k recidivě a zde by měla být léčena jako multicentrická forma (MCD) [2,4]. Multicentrická forma postihuje více skupin lymfatických uzlin nejčastěji v oblasti krku, mediastina, axily a břicha. Častěji bývá provázena symptomy, včetně zánětlivých a B symptomů (teplota nad 38 °C, noční pocení, hmotnostní úbytek více než 10 % za 6 měsíců). Nesprávná léčba MCD je běžná a vede k progresi onemocnění [2].
Typy Castlemanovy choroby ve vztahu k celkovému přežití
Typ Castlemanovy choroby určuje celkovou míru přežití [3]. U MCD je doložena vyšší úmrtnost než u mnoha běžných druhů onkologických onemocnění postihujících měkké tkáně a lymfomů, včetně kolorektálního karcinomu ve stadiu II, karcinomu prsu ve stadiu III a progresivního non‑Hodgkinova lymfomu [5]. Idiopatická MCD (iMCD) zahrnuje více než 50 % případů MCD, nemá vztah k infekci virem lidské imunodeficience/lidským herpesvirem 8 (HIV/HHV‑8) a pacienti s touto formou reagují na léčbu odlišně [2]. Non‑idiopatická MCD bývá sekundární k infekci HIV/HHV‑8 a léčí se v rámci syndromu POEMS (Polyneuropathy, Organomegaly, Endocrinopathy, Monoclonal Gammopathy, and Skin changes) [1].
 Pacienti s MCD v dlouhodobém horizontu vždy vykazovali nižší celkové přežití [6]. Retrospektivní analýza 185 HIV negativních pacientů s Castlemanovou chorobou odhalila, že pacienti s MCD mají významně horší prognózu. Pětileté celkové přežití u pacientů s UCD bylo 93,6 % ve srovnání s 51,2 % u pacientů s MCD (p < 0,001), graf 1 [6]. Starší studie zahrnovaly i pacienty s iMCD do celé skupiny MCD kvůli absenci testování na HIV/HHV‑8.
Pacienti s MCD v dlouhodobém horizontu vždy vykazovali nižší celkové přežití [6]. Retrospektivní analýza 185 HIV negativních pacientů s Castlemanovou chorobou odhalila, že pacienti s MCD mají významně horší prognózu. Pětileté celkové přežití u pacientů s UCD bylo 93,6 % ve srovnání s 51,2 % u pacientů s MCD (p < 0,001), graf 1 [6]. Starší studie zahrnovaly i pacienty s iMCD do celé skupiny MCD kvůli absenci testování na HIV/HHV‑8.
Důvodem tohoto stavu je především to, že donedávna pro iMCD neexistovala žádná diagnostická kritéria ani léčebné možnosti. Idiopatická MCD je vzácné onemocnění s roční incidencí ve Spojených státech amerických nižší než 1 000 pacientů [7]. Jde o komplexní onemocnění s mnoha podtypy a různorodým klinickým obrazem. Současný výzkum iMCD je založen na výsledcích několika publikovaných systematických studií. Nedostatek údajů z reálné praxe však ztěžuje přístup k informacím o léčebných odpovědích u pacientů mimo klinické studie [7]. Nepodávání léčby či její pozdní zahájení nebo nesprávná terapie přispívaly v minulosti ke špatným klinickým výsledkům [2].
Obtíže při diagnostice iMCD
Diagnostika iMCD je obtížná z důvodu symptomů překrývajících se s jinými malignitami a infekcemi. Mezi iMCD, jinými malignitami, autoimunitními a infekčními chorobami existuje významný klinický, histologický a imunologický přesah [8]. Zásadní je správný diagnostický postup, včetně excize celé lymfatické uzliny/uzlin pro histopatologické vyšetření [7,9]. Projevy iMCD se mohou lišit a variovat od mírných celkových flu‑like symptomů až po život ohrožující cytokinovou bouři, selhání orgánů a smrt [7,8,10].
Časté klinické příznaky a život ohrožující příznaky shrnuje předchozí článek MUDr. Écsiové [11–13].
Příznaky iMCD jsou spojeny s nadprodukcí interleukinu 6
Dysregulovaná nepřetržitá produkce interleukinu 6 (IL‑6) hraje patologickou roli (kromě jiných onemocnění) také u iMCD [14,15].
- Hypergamaglobulinemie: nadměrná stimulace B lymfocytů vede k proliferaci plazmatických buněk, které produkují protilátky odpovědné za nadprodukci protilátek [14,15].
- Renální dysfunkce: proliferace mezangiálních buněk a nadprodukce matrix jsou charakteristické rysy glomerulárních onemocnění a IL‑6 byl identifikován v depozitech matrix [15].
- Malignita: IL‑6 působí jako růstový faktor pro hybridomové a myelomové buňky.
- Angiogeneze a osteoporóza: stromální buňky kostní dřeně produkují IL‑6, jenž stimuluje receptorový aktivátor ligandu NFkB (RANKL), který je nezbytný pro diferenciaci a aktivaci osteoklastů. Tyto pochody vedou ke kostní resorpci a osteoporóze. Interleukin 6 také indukuje produkci vaskulárního endotelového růstového faktoru (VEGF), což má za následek angiogenezi a zvýšenou vaskulární permeabilitu – patologické znaky onkologických a zánětlivých lézí v synoviálních tkáních revmatoidní artritidy [15].
- Ztráta imunitní suprese: specifické efektorové Th17 lymfocyty eliminují extracelulární patogeny a IL‑6 indukovaná dominance Th17 lymfocytů nad regulačními T lymfocyty může být zodpovědná za narušení imunitní tolerance, která se podílí na rozvoji autoimunitních a zánětlivých onemocnění [15].
- Akutní fáze zánětlivé reakce: IL‑6 stimuluje hepatocyty k produkci proteinů akutní fáze, jež představují C‑reaktivní protein, sérový amyloid A, fibrinogen, hepcidin a alfa1 antichymotrypsin, a snižuje produkci fibronektinu, albuminu a transferinu [15].
- Trombocytóza: IL‑6 stimuluje megakaryocyty k produkci krevních destiček. To v důsledku vede k trombocytóze.
- Tvorba multipotentních kolonií: IL‑6 aktivuje hematopoetické kmenové buňky, což vede k tvorbě multipotentních kolonií.
Léčebný algoritmus pro iMCD
Pro nedostačené povědomí lékařů o iMCD vytvořila mezinárodní skupina odborníků Castleman Disease Collaborative Network (CDCN) léčebný algoritmus založený na nejnovějších důkazech o iMCD (na základech medicíny založené na důkazech, EBM) [7,16]. Kategorie důkazů a konsenzů byly navrženy podle kategorií National Comprehensive Cancer Network (NCCN).
Zde uvádíme jen stručný výtah, protože některé léčivé přípravky, jež mají registraci amerického Úřadu pro kontrolu potravin a léčiv (FDA), v Evropské unii schválení v indikaci léčby iMCD postrádají.
Pro dysregulaci produkce IL‑6 u iMCD je doporučena anti‑IL‑6 terapie jako léčba první volby. Siltuximab je doporučen jako první linie léčby mírné i závažné iMCD (síla důkazů 1A). Nedoporučuje se převedení na jinou anti‑IL‑6 monoklonální protilátku, pokud pacient nereaguje na siltuximab.
Tocilizumab není schválen pro léčbu iMCD, nicméně jeho podání bylo sledováno v zemích, kde siltuximab není dostupný (důkaz kategorie 2A).
Rituximab se doporučuje jako druhá linie po selhání terapie anti-IL‑6 a může být kombinován s kortikosteroidy a/nebo s imunomodulačními/imunosupresivními látkami. Může to být alternativní možnost pro některé mírné formy iMCD bez výrazné symptomatologie způsobené nadprodukcí IL‑6. Rituximab však obvykle neposkytuje dlouhodobou kontrolu iMCD a relapsy jsou běžné [17].
U pacientů se závažnou iMCD s nedostatečnou odpovědí na terapii anti-IL‑6 lze vyzkoušet kombinovanou chemoterapii. Kortikosteroidy jsou užitečnou doplňkovou terapií a dávku lze upravit podle závažnosti onemocnění, ale nedoporučují se jako samostatná léčba iMCD [16].
Benefity cílené léčby iMCD
Historické studie a nedávná analýza pacientů s iMCD léčených tradiční a necílenou terapií ukazují průměrné pětileté přežití 65 % [11,18]. Naproti tomu nedávná dlouhodobá studie u pacientů s iMCD léčených cílenou léčbou anti‑IL‑6 prokázala pětileté přežití u 96,4 % nemocných [11]. Pacienti s iMCD léčení anti‑IL‑6 monoklonální protilátkou vykazují delší přežití i delší dobu do progrese onemocnění [11,18].
 Pouhé pozorování (watch and wait), které se historicky používalo u pacientů s nezávažnou formou onemocnění, poskytuje nulovou míru dosažení kompletní odpovědi (complete response, CR) u iMCD [2,18]. Chemoterapie nebo léčba kortikosteroidy poskytly 16% podíl CR. Monoterapie kortikosteroidy není doporučena pro léčbu iMCD, ale může být užitečnou doplňkovou terapií [7,18]. Bylo prokázáno, že režimy obsahující rituximab mají 20–33% podíl CR, včetně údajů z nedávné studie z Číny. U většiny pacientů s iMCD se však rituximab jako terapie první volby nedoporučuje [2,7,17]. Terapie anti‑IL‑6 monoklonální protilátkou je doporučena jako první linie u pacientů s mírnou i závažnou iMCD a vykazuje u 43 % nemocných CR a u 85 % osob CR/stabilizaci onemocnění [7,11]. Nedávná analýza výsledků 31 pacientů ukázala progresi u všech neléčených nemocných s nezávažnou iMCD (8 z 8) a prodloužené přežití bez progrese (PFS) u nemocných léčených anti‑IL‑6 terapií [2].
Pouhé pozorování (watch and wait), které se historicky používalo u pacientů s nezávažnou formou onemocnění, poskytuje nulovou míru dosažení kompletní odpovědi (complete response, CR) u iMCD [2,18]. Chemoterapie nebo léčba kortikosteroidy poskytly 16% podíl CR. Monoterapie kortikosteroidy není doporučena pro léčbu iMCD, ale může být užitečnou doplňkovou terapií [7,18]. Bylo prokázáno, že režimy obsahující rituximab mají 20–33% podíl CR, včetně údajů z nedávné studie z Číny. U většiny pacientů s iMCD se však rituximab jako terapie první volby nedoporučuje [2,7,17]. Terapie anti‑IL‑6 monoklonální protilátkou je doporučena jako první linie u pacientů s mírnou i závažnou iMCD a vykazuje u 43 % nemocných CR a u 85 % osob CR/stabilizaci onemocnění [7,11]. Nedávná analýza výsledků 31 pacientů ukázala progresi u všech neléčených nemocných s nezávažnou iMCD (8 z 8) a prodloužené přežití bez progrese (PFS) u nemocných léčených anti‑IL‑6 terapií [2].
Proč se zlepšily klinické výsledky u iMCD
Byly zveřejněny pokyny pro diagnostiku a léčbu Castlemanovy choroby [7], včetně zařazení anti‑IL‑6 přípravků na první místo v léčebném algoritmu [2,11]. Vzácným onemocněním včetně iMCD je obecně věnováno více pozornosti a podpory [19]. Roste počet publikovaných studií a je k dispozici více dat z reálné praxe [11]. V neposlední řadě se rovněž zlepšila diagnostika onemocnění, a to včetně upřesnění kritérií léčebné odpovědi [20].
Redakčně zpracovala MUDr. Marta Šimůnková
Literatura
[1] Fajgenbaum DC. Novel insights and therapeutic approaches in idiopathic multicentric Castleman disease. Blood 2018; 132: 2323–2330.
[2] Yu L, Tu M, Cortes J, et al. Clinical and pathological characteristics of HIV‑ and HHV‑8‑negative Castleman disease. Blood 2017; 129: 1658–1668.
[3] Kaur H, Xiang Z, Kunthur A, Mehta P. Castleman Disease. Federal practitioner: for the health care professionals of the VA, DoD, and PHS 2015; 32(Suppl 7): 41s–46s.
[4] Mohan M, Meek JC, Meek ME, et al. Treatment of unresectable unicentric Castleman Disease with therapeutic embolization. Blood 2018; 132(Suppl 1): 2415.
[5] NCCN. Clinical Practice Guidelines in Oncology: B‑Cell Lymphomas, 2019. Dostupné na: https://www.nccn.org
[6] Zhang X, Rao H, Xu X, et al. Clinical characteristics and outcomes of Castleman disease: A multicenter study of 185 Chinese patients. Cancer Sci 2018; 109: 199–206.
[7] van Rhee F, Voorhees P, Dispenzieri A, et al. International, evidence‑based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood 2018; 132: 2115–2124.
[8] Fajgenbaum DC, Uldrick TS, Bagg A. International, evidence‑based consensus diagnostic criteria for HHV‑8–negative/idiopathic multicentric Castleman disease. Blood 2017; 129: 1646–1657.
[9] Navrátil M. Uzlinový syndrom, praktické poznámky k diferenciální diagnostice a diagnostickému postupu. Interní Med 2003; 1: 27.
[10] Liu AY, Nabel CS, Finkelman BS, et al. Idiopathic multicentric Castlemanʼs disease: a systematic literature review. Lancet Haematol 2016; 3: e163–e175.
[11] Sitenga J, Aird G, Ahmed A, Silberstein PT. Impact of siltuximab on patient‑related outcomes in multicentric Castlemanʼs disease. Patient Relat Outcome Meas 2018; 9: 35–41.
[12] van Rhee F, Stone K, Szmania S, et al. Castleman disease in the 21st century: An update on diagnosis, assessment, and therapy. Clin Adv Hematol Oncol 2010; 8: 486–498.
[13] El‑Osta HE, Kurzrock R. Castlemanʼs disease: from basic mechanisms to molecular therapeutics. Oncologist 2011; 16: 497–511.
[14] Yoshizaki K, Murayama S, Ito H, Koga T. The role of interleukin‑6 in Castleman disease. Hematol Oncol Clin North Am 2018; 32: 23–26.
[15] Tanaka T, Kishimoto T. The biology and medical implications of interleukin‑6. Cancer Immunol Res 2014; 2: 288–294.
[16] Castleman Disease Collaborative Network: iMCD Treatment Targets. Dostupné na: https://cdcn.org/physicians‑researchers/imcd‑treatment‑targets [navštíveno 12/2019]
[17] Dong Y, Zhang L, Nong L, et al. Effectiveness of rituximab‑containing treatment regimens in idiopathic multicentric Castleman disease. Ann Hematol 2018; 97: 1641–1647.
[18] Dispenzieri A, Armitage JO, Loe MJ, et al. The clinical spectrum of Castlemanʼs disease. Am J Hematol 2012; 87: 997–1002.
[19] Fajgenbaum DC, Ruth JR, Kelleher D, Rubenstein AH. The collaborative network approach: a new framework to accelerate Castlemanʼs disease and other rare disease research. Lancet Haematol 2016; 3: e150–152.
[20] Pierson SK, Katz L, Nabel CS, et al. Quantitative Changes in Serum Proteins Including CXCL13 Are Early Indicators of Response to Anti‑IL6 Therapy in Idiopathic Multicentric Castleman Disease. Blood 2019; 134(Suppl 1): 3599.
Úloha patologa v diagnostice Castlemanovy choroby: komplexní přístup a nutnost klinicko‑patologické spolupráce
MUDr. Kateřina Kamarádová, Ph.D.
Fingerlandův ústav 1. LF UK a FN Hradec Králové
Prezentace přednesená při příležitosti II. českého hematologického a transfuziologického sjezdu v září 2021 v Olomouci.
Už v rámci diagnostiky si musíme uvědomit, že Castlemanova choroba (CD) není jedno onemocnění. Je to skupina lézí, které pro patology mají charakteristickou morfologii, ale mají různé klinicko‑patologické podtypy, etiologii a projevy. Patolog v počátku diagnostiky obvykle neví, o kterou formu se jedná. Neví, jaký je rozsah postižení, a často to neví ani klinický pracovník. Proto se postupuje vylučováním nebo potvrzováním klinických příznaků až do stanovení diagnózy, tedy per exclusionem, což představuje určení diagnózy idiopatické multicentrické Castlemanovy choroby blíže nespecifikované (iMCD NOS).
 Castlemanova choroba představuje vzácné onemocnění, přičemž je morfologicky „vděčná“ a každý mladý patolog od začátku ví, že k charakteristickým morfologickým obrazům patří tzv. „lízátka“ a „cibule“. Co to je? Příznak cibulovitého uspořádání (onion skinning) jsou koncentrické vrstvy lymfocytů uspořádané v pláštích, které vypadají jako cibule na příčném řezu. Další krásnou morfologickou lézí je intervaskulární proliferace cév a jejich penetrace do zárodečného centra (lollipop sign), což vypadá jako lízátko na tyčince (obr. 1).
Castlemanova choroba představuje vzácné onemocnění, přičemž je morfologicky „vděčná“ a každý mladý patolog od začátku ví, že k charakteristickým morfologickým obrazům patří tzv. „lízátka“ a „cibule“. Co to je? Příznak cibulovitého uspořádání (onion skinning) jsou koncentrické vrstvy lymfocytů uspořádané v pláštích, které vypadají jako cibule na příčném řezu. Další krásnou morfologickou lézí je intervaskulární proliferace cév a jejich penetrace do zárodečného centra (lollipop sign), což vypadá jako lízátko na tyčince (obr. 1).
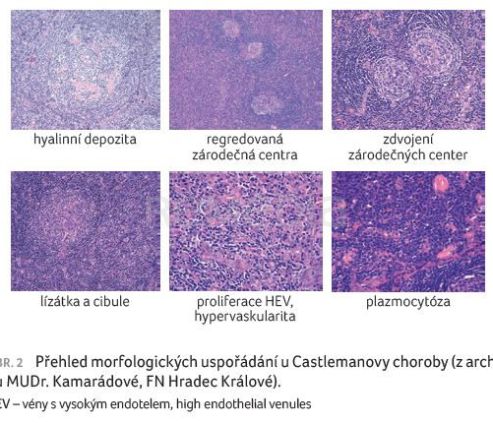 Mezi další znaky patří zdvojení zárodečných center, tzn. twinning, a u plazmocytární varianty výrazné zmnožení plazmocytů a hyperplastická zárodečná centra (obr. 2).
Mezi další znaky patří zdvojení zárodečných center, tzn. twinning, a u plazmocytární varianty výrazné zmnožení plazmocytů a hyperplastická zárodečná centra (obr. 2).
Proč vypadá Castlemanova choroba tak, jak vypadá?
Významnou roli tu hrají cytokiny, především interleukin 6 (IL‑6), ať už je produkován nádorovými plazmocyty, nebo virem aktivovaného imunitního systému. K produkci IL‑6 přispívá aktivace vaskulárního endoteliálního růstového faktoru. To znamená, že morfologie bude velice podobná u všech jednotlivých podtypů.
Castlemanova choroba očima patologa
Prvním problémem je různý stupeň vyjádření morfologických znaků v rámci jedné uzliny, a to včetně parciálního postižení uzliny nebo pouze mírně naznačeného rozvoje znaků. Proto v diagnostických kritériích iMCD jsou uvedena histopatologická kritéria pro každý morfologický znak zvlášť.
Jako minimum pro vyslovení podezření na CD je nutný grade 2–3 pro regresivní zárodečná centra nebo plazmocytóza pro plazmocytární variantu. Nicméně problémem zůstává, že v době diagnostiky není jasné, zda se jedná o unicentrickou lézi, nebo zda jde o vícečetné lokalizace uzlinového postižení.
Klinické příznaky mohou mít široké spektrum od infekcí přes autoimunity až po příznaky lymfomové, tedy B symptomy, a proto často zní popisný závěr „Castleman‑like“ změny v lymfatické uzlině s nutností klinické korelace a zpětné vazby. S rozpaky nad konečnou diagnózou se patolog setkává například v rámci „druhého čtení“ vzorků z externích pracovišť, kdy nemá klinickou zpětnou vazbu. Unicentrická CD je častěji hyalinně vaskulární a s lidským herpesvirem 8 (HHV‑8) asociovaná je spíše plazmocytární. Nicméně existuje široká šedá zóna smíšených variant. Historicky zmiňovaný plazmablastický typ je v současné době podle Světové zdravotnické organizace v kategorii germinotropní lymfoproliferativní poruchy, to znamená v kategorii mikrolymfomu.
Diagnostická kritéria pro iMCD jsou pro patology velmi přínosná, protože obsahují i návod k diferenciální diagnóze. Doporučený postup jasně definuje, co může vyloučit nebo potvrdit patolog a k čemu potřebuje klinika. Jde především o laboratorní a klinická kritéria, ale zároveň i o velkou skupinu autoimunitních onemocnění.
Diagnóza per exclusionem
Další rolí patologa je postupné vylučování lézí, které jsou v diagnostických kritériích uvedena jako „k vyloučení“. Patolog si může pomoci imunohistochemickým vyšetřením, in situ hybridizací, případně molekulárními metodami.
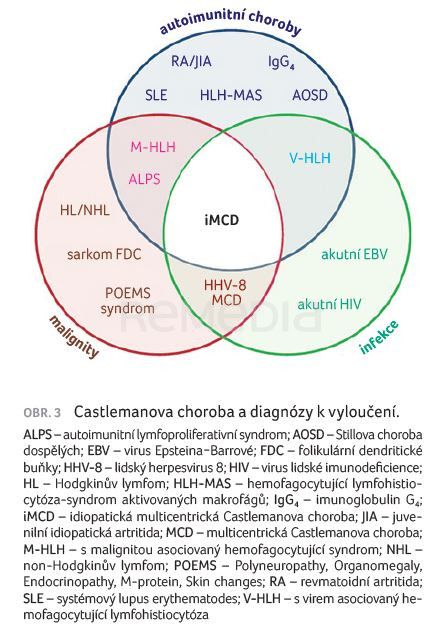 Spektrum lézí k vyloučení je opravdu široké (obr. 3).
Spektrum lézí k vyloučení je opravdu široké (obr. 3).
Jde například o:
- autoimunitní onemocnění (systémový lupus erythematodes, revmatoidní artritida/JIA, s IgG4 asociovaná choroba, Stillova choroba dospělých, ALPS syndrom, hemofagocytární lymfohistocytóza),
- infekce (EBV, HIV),
- hematologické malignity (lymfoproliferace – HL/NHL, MM, POEMS syndrom s PC neoplazií, FDC sarkom).
Z infekcí je virus Epsteina–Barrové (EBV) na prvním místě. Nejde však o akutní EBV, tedy infekční mononukleózu, která je spojena spíše s folikulární hyperplazií u mladších osob, ale především o chronickou aktivní EBV a o lymfoproliferace asociované s EBV, které mohou s CD sdílet i morfologii. V tomto případě je nejnebezpečnější angioimunoplastický T lymfom, (AITL, pattern 1), pro který jsou charakteristická drobná regredovaná zárodečná centra. Ve skutečnosti to však nejsou zárodečná centra, ale už proliferace nádorových T lymfocytů.
IgG4 asociovaná choroba je fibroinflamatorní onemocnění, které postihuje různé orgány, ale i lymfatické uzliny, kde má až pět lymfatických podtypů, a morfologie typu CD je jednou z nich. Nejspecifičtější vzhled představuje interfolikulární expanze a inflamatorní pseudotumor‑like verze. IgG4 asociovaná choroba byla dříve dávána do souvislosti s CD, průkaz však chybí. U CD pomůže v diagnostice například elevace hodnot IgA v séru nebo imunohistochemická detekce IgA v plazmocytech.
Další kategorií v diferenciální diagnostice jsou lymfoproliferace.
Castleman‑like změny v lymfomech
Patology může zmást Castleman‑like morfologie v lymfomech, která je považována za raritní. Je však otázkou, zda to nejsou léze, které jsou diagnostikovány z preparátů obarvených hematoxylinem‑eosinem jako Castleman‑like změny, a není u nich doplněno imunologické vyšetření. Nejčastěji je popsána v lézích, které vycházejí z folikulů nebo ze zárodečného centra. Jedná se o non‑hodgkinské lymfomy, ale i o Hodgkinův lymfom, u kterého je rovněž popsána koexistence v jedné lymfatické uzlině. Morfologie může být ovlivněna expresí IL‑6 v nádorových buňkách Reedové–Sternberga.
Pacienti s CD mohou mít i sekundární malignity. Jednak jsou ohroženi větším rizikem rozvoje lymfoproliferativních onemocnění, a to především pacienti s HHV‑8 asociovanou CD, ale i s UCD, u níž jde zejména o sarkom z folikulárních dendritických buněk. Jednak může u pacientů s UCD i MCD jít o hodgkinské i non‑hodgkinské lymfomy.
Samostatnou kapitolou jsou již zmíněné HHV‑8 pozitivní lymfoproliferace. Tam spadá jak difuzní velkobuněčný B lymfom, tak i germinotropní lymfoproliferativní porucha, u které můžeme vidět monotypickou expresi lehkých řetězců imunoglobulinů (kappa nebo lambda). Často je přítomen pozitivní průkaz EBV, ale na rozdíl od rozvinutého lymfomu bývají ještě v molekulárním vyšetření polymerázovou řetězovou reakcí (PCR) polyklonální nebo oligoklonální imunoglobuliny.
Role patologa v diagnostice Castlemanovy choroby
Patolog musí znát spektrum morfologie u UCD i MCD. Musí si být vědom, že diagnózu CD nelze stanovit pouze z jednoho preparátu obarveného hematoxylinem‑eosinem. Musí vědět, že pro detekci diagnostických i vylučujících kritérií musí použít alespoň základní imunohistochemické vyšetření, případně další metody jako in situ hybridizaci a PCR, a mít znalost širokého spektra morfologie diferenciální diagnostiky. Vznikne‑li podezření již na preparátech se základním barvením po vyloučení lymfomu apod., pak je žádoucí komunikace s klinikem a doplnění klinických údajů v průběhu diagnostického algoritmu. Proto je vedle mikroskopu druhým nejdůležitějším nástrojem patologa telefon.
Redakčně zpracovala MUDr. Marta Šimůnková, kráceno
Od vstupu siltuximabu jsme neztratili jediného pacienta
 Rumunsko je vnímáno jako země, jejíž zdravotnictví a přístup inovativních přípravků nedosahují standardů Evropské unie. Existují však léky, které toto pravidlo nepotvrzují. Konkrétně jde o siltuximab (Sylvant), který je v Rumunsku hrazen z veřejného zdravotního pojištění a pro diagnostikované pacienty s idiopatickou multicentrickou Castlemanovou chorobou (iMCD) je dostupný. To říká v rozhovoru pro časopis Medicína po promoci hematoonkolog Ciprian Tomuleasa z Ion Chiricuta Oncology Institute v Cluj Napoca.
Rumunsko je vnímáno jako země, jejíž zdravotnictví a přístup inovativních přípravků nedosahují standardů Evropské unie. Existují však léky, které toto pravidlo nepotvrzují. Konkrétně jde o siltuximab (Sylvant), který je v Rumunsku hrazen z veřejného zdravotního pojištění a pro diagnostikované pacienty s idiopatickou multicentrickou Castlemanovou chorobou (iMCD) je dostupný. To říká v rozhovoru pro časopis Medicína po promoci hematoonkolog Ciprian Tomuleasa z Ion Chiricuta Oncology Institute v Cluj Napoca.
- Prosím, mohl byste představit kliniku, kde své pacienty léčíte?
V současné době léčíme pacienty s Castlemanovou chorobou na hematologickém oddělení v Ion Chiricuta Oncology Institute v Cluj Napoca, v jednom z nejvýznamnějších referenčních center v onkologii v Rumunsku.
- Jak dlouho se věnujete medicíně a léčbě pacientů s idiopatickou multicentrickou Castlemanovou chorobou?
Jsem ošetřujícím lékařem v klinické hematologii sedm let a již pět let léčím pacienty s Castlemanovou chorobou.
- Jaká je u vás situace s diagnostikou Castlemanovy choroby?
Mnoho pacientů je diagnostikováno pozdě; toto onemocnění je obecně nedostatečně diagnostikováno. Ti, kteří jsou diagnostikováni, dostávají nyní vynikající léčbu s neuvěřitelně dobrými výsledky.
- V EU existuje velmi málo studií prevalence iMCD. Jak tomu je v Rumunsku?
V současné době u nás neprobíhá žádné klinické hodnocení u Castlemanovy choroby, i když bychom rádi brzy zahájili akademickou studii.
- Jak stanovujete diagnózu iMCD?
Diagnóza je většinou založena na poznatcích zkušených patologů. Naštěstí máme v Cluj Napoca vynikající histopatology.
- Jak spolupracujete s patology, případně s kterými dalšími lékaři, při stanovení diagnózy?
Je to spolupráce, která musí fungovat nejen u Castlemana, ale je tomu tak u všech hematologických poruch s cílem, aby se pro tyto pacienty nastavila nejlepší dostupná terapie.
- Jaké léky jste používali dříve, než se k vám dostal siltuximab?
Před siltuximabem byla terapie většinou založena na klasické chemoterapii s omezenými výsledky. Výsledky léčby Castlemanovy choroby byly tehdy špatné.
- Jak změnil situaci pacientů s iMCD příchod siltuximabu?
Siltuximab je molekula, která skutečně změnila prognózu těchto pacientů. Od zavedení siltuximabu do léčby iMCD se situace dramaticky posunula a můžeme s hrdostí říci, že od schválení jeho úhrady jsme ještě neztratili jediného pacienta.
- Kolik pacientů s iMCD je ve vaší péči a jak se jejich onemocnění vyvíjí při současné léčbě?
V současné době jsme referenčním centrem pro náš region a počet našich diagnostikovaných pacientů s Castlemanovou chorobou stoupá.
- Je siltuximab v Rumunsku hrazen ze zdravotního pojištění?
Ano, máme velké štěstí, že lento léčivý přípravek je hrazený.
- Dostávají siltuximab všichni pacienti, pro které je indikován?
Ano, pokud jsou splněna všechna kritéria pro zahájení terapie.
Medicína po promoci 3/2023
Siltuximab
MUDr. Tomáš Doležal, Ph.D.1; prof. MUDr. Zdeněk Adam, CSc.2
1 iHETA, Praha; 2 Interní hematologická a onkologická klinika FN a LF MU, Brno
Souhrn
Doležal T, Adam Z. Siltuximab. Farmakoterapie 2021; 17: 475–483.
Castlemanova choroba (CD) je vzácné, heterogenní onemocnění postihující lymfatické uzliny v jedné či více oblastech. Multicentrická CD (MCD) je charakterizována generalizovanou lymfadenopatií, hepatosplenomegalií a systémovými příznaky, jako jsou horečka, únava, hubnutí, nechutenství či noční pocení, které vznikají na podkladě dysregulace a trvalé nadprodukce interleukinu 6 (IL‑6) B lymfocyty zárodečných center hyperplastických lymfatických uzlin. Poznatek o klíčové úloze IL‑6 v patogenezi MCD vedl k testování siltuximabu, chimérické monoklonální anti‑IL‑6 protilátky. V klinických studiích u pacientů s idiopatickou MCD byla pozorována významná účinnost siltuximabu s ohledem na nádorovou i symptomatickou léčebnou odpověď, přičemž bezpečnostní profil siltuximabu byl velmi dobrý. Na základě těchto výsledků byl siltuximab registrován pro terapii dospělých pacientů s multicentrickou formou Castlemanovy choroby a negativitou HIV a HHV‑8.
Klíčová slova: Castlemanova choroba – HHV‑8 – interleukin 6 – siltuximab.
Summary
Dolezal T, Adam Z. Siltuximab. Farmakoterapie 2021; 17: 475–483.
Castlemanʼs disease (CD) is a rare and heterogeneous disease affecting the lymph nodes in one or more areas. Multicentric CD (MCD) is characterized by generalized lymphadenopathy, hepatosplenomegaly and systemic symptoms such as fever, fatigue, weight loss, loss of appetite or night sweats, which result from dysregulation and persistent overproduction of interleukin‑6 (IL‑6) by B‑cells of germinal centres of hyperplastic lymph nodes. Understanding the key role of IL‑6 in the pathogenesis of MCD has led to the testing of siltuximab, a chimeric monoclonal anti‑IL‑6 antibody. In clinical trials in patients with idiopathic MCD, significant efficacy of siltuximab was observed in terms of both tumour and symptomatic response and the safety profile of siltuximab was very favourable. Based on these results, siltuximab has been authorized for the treatment of adult patients with multicentric Castlemanʼs disease who are HIV and HHV‑8 negative.
Key words: Castlemanʼs disease – HHV‑8 – interleukin‑6 – siltuximab.
Úvod
Castlemanova choroba (CD) je vzácně se vyskytující heterogenní lymfoproliferativní onemocnění, jehož patogeneze dosud není zcela známa. Důležitou úlohu při jejím vzniku hraje interleukin 6 (IL‑6) (bylo zjištěno, že u myší indukuje IL‑6 stav podobný CD) [1], významný prozánětlivý cytokin, který mj. reguluje i hematopoezu (stimulace vyzrávání B lymfocytů). Při vzniku CD se mohou uplatňovat též další cytokiny jako IL‑1, IL‑2 či vaskulární endoteliální růstový faktor (VEGF) [2]. Část případů CD vzniká na podkladě infekce HHV‑8 (tento virus produkuje analog IL‑6, který má podobné účinky jako lidský IL‑6 a rovněž zvyšuje koncentraci lidského IL‑6), nalezena byla rovněž asociace mezi CD a infekcí HIV či HCV [3]. Virus HHV‑8 způsobuje CD u imunokompromitovaných pacientů, jako jsou právě osoby s infekcí HIV [2].
Z klinického hlediska rozlišujeme unicentrickou a multicentrickou formu CD, z histologického hlediska pak hyalinně vaskulární, plazmocelulární a smíšenou variantu nemoci. Tyto typy se mezi sebou liší nejen klinickými projevy, ale i způsobem léčby a prognózou. Při unicentrické CD (obvykle hyalinně vaskulární) se jedná o postižení jedné uzlinové oblasti, kdy dochází k pomalému, benignímu, obvykle nebolestivému zvětšování lymfatické uzliny (LU). Onemocnění bývá asymptomatické, může však být provázeno příznaky plynoucími z komprese okolních struktur. Unicentrická CD se nejčastěji objevuje u osob ve věku 20–35 let a nejčastěji bývají postiženy LU v oblasti mediastina, břicha či periferní LU. Unicentrická forma CD je vyléčitelná pomocí kompletní chirurgické excize postižených LU a prognóza onemocnění je velmi dobrá [3].
Naproti tomu multicentrická forma CD (obvykle plazmocelulární) je méně častá (představuje asi 10 %, dle jiných zdrojů až 30 % případů CD) a vyskytuje se u starších osob (medián věku 48–57 let) [3,4]. Je charakterizována generalizovanou lymfadenopatií, hepatosplenomegalií a systémovými příznaky (horečka, únava, hubnutí, nechutenství, noční poty), které vznikají na podkladě dysregulace a trvalé nadprodukce IL‑6 B lymfocyty zárodečných center hyperplastických LU (nadprodukce IL‑6 je zřejmě zodpovědná i za špatný nutriční stav pacientů s multicentrickou CD a může se uplatňovat i při rozvoji kachexie a chronického zánětu) [5]. Laboratorně je nacházena mikrocytární anémie (IL‑6 zvyšuje tvorbu hepcidinu, který blokuje uvolňování železa z jaterních makrofágů, a zároveň snižuje absorpci železa ve střevě, čímž se snižuje přísun železa do vyvíjejících se erytrocytů v kostní dřeni) [6], dále hypoalbuminemie, hypocholesterolemie, hypergamaglobulinemie (na podkladě polyklonálního zvýšení všech imunoglobulinů, nejvíce však typu IgG) a elevace proteinů akutní fáze. Multicentrická forma CD (MCD) mívá progresivní klinický průběh, který může nabývat různého stupně agresivity od mírného průběhu, označovaného jako „flu‑like“, až po agresivní průběh, označovaný jako „sepsis‑like“ [3].
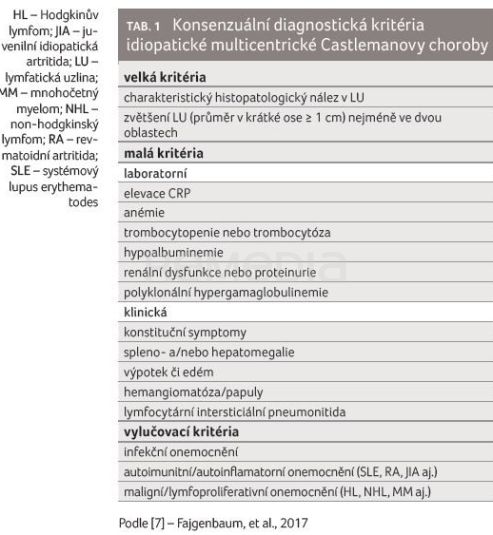 V případech, kdy se neprokáže HHV‑8 ani HIV, hovoříme o tzv. idiopatické MCD (iMCD) [8]. Diagnóza iMCD je založena na splnění velkých kritérií a nejméně dvou z malých kritérií, zároveň musí být vyloučeno infekční, maligní či autoimunitní onemocnění, které by mohlo napodobovat iMCD (tab. 1) [7].
V případech, kdy se neprokáže HHV‑8 ani HIV, hovoříme o tzv. idiopatické MCD (iMCD) [8]. Diagnóza iMCD je založena na splnění velkých kritérií a nejméně dvou z malých kritérií, zároveň musí být vyloučeno infekční, maligní či autoimunitní onemocnění, které by mohlo napodobovat iMCD (tab. 1) [7].
Léčba iMCD se odvíjí od závažnosti onemocnění. U pacientů s HHV‑8 negativní MCD se předpokládá, že hlavním patogenetickým faktorem je IL‑6, u pacientů s nezávažnou iMCD je proto dle aktuálních mezinárodních konsenzuálních doporučení v 1. linii indikována terapie cílená proti IL‑6 (siltuximab či tocilizumab) [9]. K té může být za účelem zvýšení kontroly onemocnění přidána léčba kortikosteroidy. Nejvyšší přínos z léčby cílené na IL‑6 lze očekávat u pacientů s výraznými zánětlivými příznaky.
Před registrací siltuximabu v indikaci Castlemanovy choroby americkým úřadem FDA a evropským úřadem EMA byl uváděn v literatuře jako lék 1. linie rituximab, jehož účinnost byla popsána v četných rozborech jednotlivých případů anebo skupin pacientů. S rituximabem proběhly registrační studie u nejčastějších typů B lymfomů (folikulární lymfom, difuzní velkobuněčný B lymfom atd.), v nichž se osvědčil. Proto je podáván i u dalších onemocnění, v jejichž patogenezi dominuje proliferace B lymfocytů, ať již klonální (Waldenströmova makroglobulinemie), nebo polyklonální (onemocnění asociované s IgG4 a četné další nemoci ze skupiny autoimunitních chorob), takzvaně „off‑label“ neboli na § 16 se schválením plátců zdravotní péče. V současnosti jsou na trhu již biosimilars odvozené od rituximabu od dalších firem, a nelze tedy očekávat, že by vznikaly oficiální registrační studie s rituximabem pro další diagnózy.
Co se týká porovnání účinnosti siltuximabu a rituximabu v léčbě CD, někteří autoři uvádějí, že účinnější je siltuximab [8]. Toto tvrzení však není podloženo prospektivní randomizovanou srovnávací klinickou studií, nýbrž vychází z porovnání výsledků některých observačních studií.
V případě, že je v rámci 1. linie podán siltuximab, lze rituximab využít ve 2. linii léčby po terapii anti‑IL‑6, přičemž je možné jej kombinovat s kortikosteroidy, případně s cytostatiky, která se používají pro léčbu maligních lymfomů. V literatuře je také více popisů případů dokumentujících dobrou účinnost léků ze skupiny IMID (immunomodulatory drugs), konkrétně thalidomidu a lenalidomidu, zatím však chybí publikace hodnotící nejnovější IMID – pomalidomid – v této indikaci. V několika případech rezistentních na předchozí terapii bylo dosaženo léčebné odpovědi při využití anakinry (anti‑IL‑1) [3,9].
U pacientů se závažnou formou iMCD (sepsis‑like) by měla být zahájena intenzivní léčba. Publikované guidelines doporučují vysokodávkované kortikosteroidy v kombinaci se siltuximabem zpočátku podávaným v týdenních intervalech; po zvládnutí akutní ataky následuje postupné snižování dávek kortikosteroidů a pokračování siltuximabu ve standardních intervalech. Dříve byla v této indikaci využívána kombinovaná léčba anti‑CD20 monoklonální protilátkou (rituximabem) a chemoterapií, tj. léčebné režimy používané v terapii B lymfomů [9].
Charakteristika a mechanismus účinku siltuximabu
Siltuximab je chimérická (lidská/myší) monoklonální protilátka IgG1κ, která se s vysokou afinitou a specificitou váže na lidský IL‑6. Siltuximab brání vazbě IL‑6 na solubilní i membránově vázané receptory pro IL‑6 (IL‑6R) a inhibuje tvorbu hexamerního signálního komplexu s proteinem gp130 na povrchu buňky. Interleukin 6 je pleiotropní prozánětlivý cytokin produkovaný různými typy buněk (T a B lymfocyty, monocyty, fibroblasty i maligními buňkami), který se uplatňuje při imunitní odpovědi (podílí se na indukci sekrece imunoglobulinů), reakcích akutní fáze (podporuje jaterní syntézu proteinů akutní fáze), hematopoeze (stimuluje proliferaci a diferenciaci hematopoetických buněk) a kostním metabolismu [10,11]. Nadprodukce IL‑6 u chronických zánětlivých onemocnění a malignit je spojována s anémií a kachexií (viz výše) a soudí se, že tento cytokin hraje ústřední roli při stimulaci proliferace plazmatických buněk a při systémových projevech u pacientů s MCD [4,5,11].
Farmakokinetické vlastnosti
Po prvním podání siltuximabu v rozmezí dávek 0,9–15 mg/kg se hodnoty plochy pod křivkou koncentrací v čase (AUC) a maximální sérové koncentrace (cmax) zvyšovaly úměrně dávce a hodnota clearance (CL) byla nezávislá na dávce. Po jednorázové aplikaci dávky při doporučeném dávkovacím režimu (11 mg/kg každé 3 týdny) činila hodnota clearance 3,54 ± 0,44 ml/kg/den a biologický poločas eliminace byl 16,3 ± 4,2 dny. Po opakovaném podávání doporučených dávek byla zjištěna časově neměnná clearance siltuximabu a středně vysoká systémová kumulace (kumulační index 1,7). V souladu s biologickým poločasem po podání první dávky bylo dosaženo rovnovážného stavu sérové koncentrace do šesté infuze (interval každé 3 týdny) s průměrnou (± SD) maximální koncentrací 332 ± 139 μg/ml a minimální koncentrací 84 ± 66 μg/ml [11].
Clearance siltuximabu nebyla ovlivněna pohlavím, etnickým původem ani věkem (farmakokinetika siltuximabu u pacientů starších 65 let v porovnání s pacienty ve věku 65 let a mladšími byla podobná), zvyšovala se však se stoupající tělesnou hmotností (siltuximab je dávkován podle tělesné hmotnosti). Studie, které by hodnotily vliv poškození ledvin nebo jater na farmakokinetiku siltuximabu, nebyly provedeny. Nicméně u pacientů s výchozí vypočtenou clearance kreatininu ≥ 12 ml/min nebyl pozorován žádný významný vliv na farmakokinetiku siltuximabu; podobně u pacientů s výchozí hodnotou ALT odpovídající až 3,7násobku horní hranice normy, výchozí koncentrací albuminu 15–58 g/l a výchozí koncentrací bilirubinu 1,7–42,8 mg/dl nebyl pozorován žádný významný vliv na farmakokinetiku siltuximabu [11].
Lékové interakce
Interleukin 6 snižuje aktivitu cytochromu P450 (CYP450), v důsledku vazby siltuximabu na IL‑6 tak může dojít ke zvýšení metabolismu substrátů CYP450. Při zahájení nebo ukončení léčby siltuximabem u pacientů užívajících léčiva, která jsou metabolizována prostřednictvím CYP450 a mají úzký terapeutický index, se proto doporučuje monitorování účinku (např. u warfarinu) nebo koncentrace léčiva (např. u cyklosporinu nebo theofylinu) s případnou úpravou dávkování souběžně užívaných léčiv. Účinek siltuximabu na aktivitu CYP450 může přetrvávat ještě několik týdnů po ukončení léčby [11].
Účinnost v klinických studiích
Aktivita siltuximabu v monoterapii byla dokumentována v otevřené dávkové 7kohortové studii fáze I, do které bylo zařazeno 67 pacientů, z toho 37 nemocných s CD. V souboru 36 hodnotitelných pacientů byla zaznamenána kompletní odpověď (CR) u jednoho pacienta, částečná odpověď (PR) u 11, nepotvrzená PR u tří a stabilizace onemocnění u 20 pacientů; k progresi onemocnění došlo u jediného pacienta. Dosahování léčebné odpovědi bylo podobné u všech tří histologických subtypů MCD. Mediánu trvání léčebné odpovědi (DOR) ani mediánu doby do progrese (TTP) nebylo dosaženo. Úmrtí bylo při mediánu délky sledování 2,4 roku zaznamenáno pouze u 8 % pacientů [12]. Van Rhee et al. samostatně publikovali výsledky 23 pacientů léčených v této studii v dávkách 6, 9 či 12 mg/kg. Všichni tito pacienti s výjimkou jednoho měli MCD, z toho 10 mělo hyalinně vaskulární variantu a 12 plazmocelulární variantu (jeden pacient měl smíšenou variantu); všichni pacienti byli negativní stran infekce HIV i HHV‑8. Objektivní odpověď byla zjištěna u 12 pacientů (z toho u jednoho CR a u 11 PR), častěji přitom byla zaznamenána u nemocných léčených dávkou 12 mg/kg než u pacientů léčených nižšími dávkami (73 % vs. 33 %). Medián doby do radiologické PR byl 185 dní. Léčba siltuximabem byla provázena zmírněním klinických příznaků i zlepšením laboratorních parametrů (pokles koncentrace proteinů akutní fáze jako CRP a fibrinogenu, zvýšení hodnoty hemoglobinu, snížení sedimentace) a umožnila přerušení léčby současně podávanými kortikosteroidy [13].
 Na uvedenou studii fáze I navázala multicentrická randomizovaná dvojitě zaslepená placebem kontrolovaná studie fáze II, do které byli zařazeni dospělí HIV a HHV‑8 negativní pacienti s MCD ve výkonnostním stavu (PS) dle ECOG 0–2 [4]. Ti byli v poměru 2 : 1 náhodně přiřazeni k terapii siltuximabem v dávce 11 mg/kg každé 3 týdny v i.v. infuzi, nebo k podávání placeba, zároveň jim byla poskytnuta nejlepší podpůrná péče (BSC). Randomizace byla stratifikována na základě užívání kortikosteroidů při vstupu do studie. Primárním sledovaným ukazatelem byla nádorová odpověď provázená zmírněním či stabilizací symptomů trvající alespoň 18 týdnů, k sekundárním sledovaným ukazatelům patřily nádorová odpověď, trvání nádorové a symptomatické odpovědi, doba do selhání léčby, četnost přerušení léčby kortikosteroidy, celkové přežití a výsledky udávané pacienty (PRO) [4]. Do studie bylo zařazeno celkem 79 pacientů, z nichž 53 bylo randomizováno k léčbě siltuximabem a 26 k užívání placeba. Medián věku pacientů byl 47, resp. 48 let, v souboru převažovali muži (57 %, resp. 85 %), běloši (36 %, resp. 46 %) a Asiaté (51 %, resp. 42 %). Hyalinně vaskulární, plazmocelulární a smíšenou variantu MCD mělo 34 %, 25 % a 42 % pacientů ze skupiny siltuximabu, resp. 31 %, 19 % a 50 % pacientů z placebové skupiny. Kortikosteroidy konkomitantně užívalo 25 %, resp. 35 % pacientů. Výchozí průměrná koncentrace IL‑6, CRP, a zejména sedimentace byly výrazně vyšší a hodnota hemoglobinu byla nižší ve skupině se siltuximabem v porovnání s placebovou skupinou. Nejčastějšími symptomy byly únava (86 %), malátnost (61 %), noční pocení (52 %), periferní senzorická neuropatie (38 %), nechutenství (37 %), pruritus (37 %), dušnost (35 %) nebo otoky končetin (30 %). Medián trvání léčby siltuximabem byl 375 dní, medián doby podávání placeba byl 152 dní. Medián délky sledování dosáhl 422 dní; v době analýzy setrvávalo na léčbě siltuximabem 59 % pacientů a 23 % nadále užívalo placebo [4].
Na uvedenou studii fáze I navázala multicentrická randomizovaná dvojitě zaslepená placebem kontrolovaná studie fáze II, do které byli zařazeni dospělí HIV a HHV‑8 negativní pacienti s MCD ve výkonnostním stavu (PS) dle ECOG 0–2 [4]. Ti byli v poměru 2 : 1 náhodně přiřazeni k terapii siltuximabem v dávce 11 mg/kg každé 3 týdny v i.v. infuzi, nebo k podávání placeba, zároveň jim byla poskytnuta nejlepší podpůrná péče (BSC). Randomizace byla stratifikována na základě užívání kortikosteroidů při vstupu do studie. Primárním sledovaným ukazatelem byla nádorová odpověď provázená zmírněním či stabilizací symptomů trvající alespoň 18 týdnů, k sekundárním sledovaným ukazatelům patřily nádorová odpověď, trvání nádorové a symptomatické odpovědi, doba do selhání léčby, četnost přerušení léčby kortikosteroidy, celkové přežití a výsledky udávané pacienty (PRO) [4]. Do studie bylo zařazeno celkem 79 pacientů, z nichž 53 bylo randomizováno k léčbě siltuximabem a 26 k užívání placeba. Medián věku pacientů byl 47, resp. 48 let, v souboru převažovali muži (57 %, resp. 85 %), běloši (36 %, resp. 46 %) a Asiaté (51 %, resp. 42 %). Hyalinně vaskulární, plazmocelulární a smíšenou variantu MCD mělo 34 %, 25 % a 42 % pacientů ze skupiny siltuximabu, resp. 31 %, 19 % a 50 % pacientů z placebové skupiny. Kortikosteroidy konkomitantně užívalo 25 %, resp. 35 % pacientů. Výchozí průměrná koncentrace IL‑6, CRP, a zejména sedimentace byly výrazně vyšší a hodnota hemoglobinu byla nižší ve skupině se siltuximabem v porovnání s placebovou skupinou. Nejčastějšími symptomy byly únava (86 %), malátnost (61 %), noční pocení (52 %), periferní senzorická neuropatie (38 %), nechutenství (37 %), pruritus (37 %), dušnost (35 %) nebo otoky končetin (30 %). Medián trvání léčby siltuximabem byl 375 dní, medián doby podávání placeba byl 152 dní. Medián délky sledování dosáhl 422 dní; v době analýzy setrvávalo na léčbě siltuximabem 59 % pacientů a 23 % nadále užívalo placebo [4].
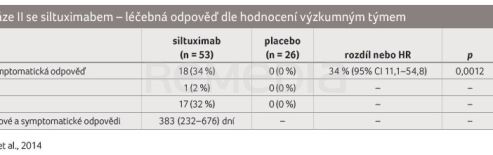
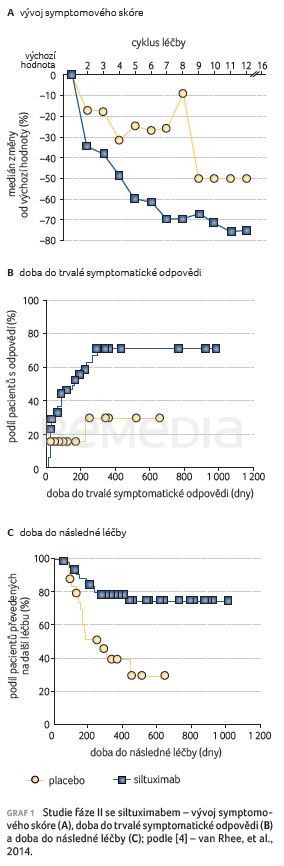 Trvalá nádorová a symptomatická odpověď byla dokumentována u 34 % pacientů léčených siltuximabem (1× CR a 17× PR) vs. u 0 % pacientů užívajících placebo (HR 34,0 %; 95% CI 11,1–54,8; p = 0,0012) s mediánem trvání odpovědi 383 dní (tab. 2). V době analýzy byla zaznamenána progrese pouze u jednoho z 18 respondérů. Mediánu doby do selhání léčby ve skupině siltuximabu nebylo dosaženo (95% CI 378–nedosaženo), zatímco v placebové skupině činil 134 dní (95% CI 85–nedosaženo; p = 0,0084). Z důvodu selhání léčby přerušilo terapii siltuximabem 30 % a podávání placeba 54 % pacientů. Symptomové skóre s každým cyklem léčby klesalo, výrazněji ve skupině se siltuximabem (graf 1A). Trvalá symptomatická odpověď byla statisticky významně častější při léčbě siltuximabem než při podávání placeba (p = 0,0018), podobně jako trvalá kompletní symptomatická odpověď (p = 0,0037); medián doby do trvalé symptomatické odpovědi byl při léčbě siltuximabem 170 dní, zatímco při podávání placeba nebyl dosažen (p = 0,0288) (graf 1B). Mediánu doby do následné léčby ve skupině se siltuximabem nebylo dosaženo vs. v placebové skupině činil 280 dní (p = 0,0013) (graf 1C) [4].
Trvalá nádorová a symptomatická odpověď byla dokumentována u 34 % pacientů léčených siltuximabem (1× CR a 17× PR) vs. u 0 % pacientů užívajících placebo (HR 34,0 %; 95% CI 11,1–54,8; p = 0,0012) s mediánem trvání odpovědi 383 dní (tab. 2). V době analýzy byla zaznamenána progrese pouze u jednoho z 18 respondérů. Mediánu doby do selhání léčby ve skupině siltuximabu nebylo dosaženo (95% CI 378–nedosaženo), zatímco v placebové skupině činil 134 dní (95% CI 85–nedosaženo; p = 0,0084). Z důvodu selhání léčby přerušilo terapii siltuximabem 30 % a podávání placeba 54 % pacientů. Symptomové skóre s každým cyklem léčby klesalo, výrazněji ve skupině se siltuximabem (graf 1A). Trvalá symptomatická odpověď byla statisticky významně častější při léčbě siltuximabem než při podávání placeba (p = 0,0018), podobně jako trvalá kompletní symptomatická odpověď (p = 0,0037); medián doby do trvalé symptomatické odpovědi byl při léčbě siltuximabem 170 dní, zatímco při podávání placeba nebyl dosažen (p = 0,0288) (graf 1B). Mediánu doby do následné léčby ve skupině se siltuximabem nebylo dosaženo vs. v placebové skupině činil 280 dní (p = 0,0013) (graf 1C) [4].
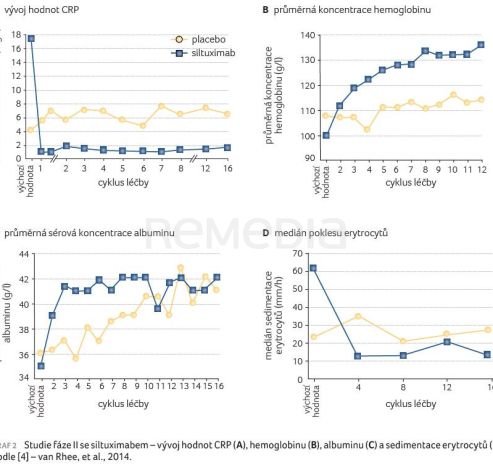 Léčba siltuximabem v porovnání s placebem byla provázena také výraznějším zlepšením sledovaných laboratorních parametrů. Vzhledem k dobré korelaci mezi hodnotou IL‑6 a koncentrací CRP a interferenci s měřením hladin IL‑6 při léčbě siltuximabem byly jako ukazatel inhibice aktivity IL‑6 sledovány hodnoty CRP. Při terapii siltuximabem byl pozorován rychlý pokles koncentrace CRP, zatímco u pacientů užívajících placebo zůstaly koncentrace CRP beze změn (graf 2A). Ve skupině se siltuximabem v porovnání s placebovou skupinou bylo pozorováno také výraznější zvýšení hodnoty hemoglobinu (graf 2B), rychlejší vzestup koncentrace albuminu (graf 2C) a pokles sedimentace erytrocytů (graf 2D) i koncentrace fibrinogenu. U pacientů léčených siltuximabem byl zjištěn rozdíl ve výchozí hodnotě CRP (nikoliv však IL‑6) s ohledem na dosahování trvalé nádorové a symptomatické odpovědi (medián hodnoty CRP u pacientů s odpovědí činil 34,50 mg/dl vs. u pacientů bez odpovědi byl 6,32 mg/l; p = 0,022) i nejlepší nádorové odpovědi (medián hodnoty CRP 38,00 mg/dl vs. 5,09 mg/l; p = 0,0118), výchozí hodnota CRP, která by predikovala dosažení odpovědi, však nebyla identifikována [4].
Léčba siltuximabem v porovnání s placebem byla provázena také výraznějším zlepšením sledovaných laboratorních parametrů. Vzhledem k dobré korelaci mezi hodnotou IL‑6 a koncentrací CRP a interferenci s měřením hladin IL‑6 při léčbě siltuximabem byly jako ukazatel inhibice aktivity IL‑6 sledovány hodnoty CRP. Při terapii siltuximabem byl pozorován rychlý pokles koncentrace CRP, zatímco u pacientů užívajících placebo zůstaly koncentrace CRP beze změn (graf 2A). Ve skupině se siltuximabem v porovnání s placebovou skupinou bylo pozorováno také výraznější zvýšení hodnoty hemoglobinu (graf 2B), rychlejší vzestup koncentrace albuminu (graf 2C) a pokles sedimentace erytrocytů (graf 2D) i koncentrace fibrinogenu. U pacientů léčených siltuximabem byl zjištěn rozdíl ve výchozí hodnotě CRP (nikoliv však IL‑6) s ohledem na dosahování trvalé nádorové a symptomatické odpovědi (medián hodnoty CRP u pacientů s odpovědí činil 34,50 mg/dl vs. u pacientů bez odpovědi byl 6,32 mg/l; p = 0,022) i nejlepší nádorové odpovědi (medián hodnoty CRP 38,00 mg/dl vs. 5,09 mg/l; p = 0,0118), výchozí hodnota CRP, která by predikovala dosažení odpovědi, však nebyla identifikována [4].
Recentně publikovaná analýza studie fáze II ukázala, že odpověď na léčbu siltuximabem je podobná u předléčených i u nově diagnostikovaných pacientů s iMCD – trvalá nádorová a symptomatická odpověď byla dokumentována u 34,5 %, resp. 33,3 % pacientů léčených siltuximabem [14].
Bezpečnost a snášenlivost
Ve studii fáze II byl výskyt nežádoucích účinků (NÚ) jakéhokoliv stupně při terapii siltuximabem podobný jako při podávání placeba (100 % vs. 96 %), a to navzdory tomu, že délka terapie siltuximabem byla více než dvojnásobná v porovnání s délkou užívání placeba. Výskyt NÚ stupně ≥ 3 byl nižší při terapii siltuximabem (47 % vs. 54 %), podobně také výskyt NÚ vedoucích k přerušení léčby (23 % vs. 38 %). Nejčastějšími NÚ jakéhokoliv stupně při terapii siltuximabem byly pruritus, infekce horních cest dýchacích, únava, makulopapulózní rash či periferní otoky; nejčastější NÚ stupně ≥ 3 při léčbě siltuximabem pak zahrnovaly únavu a noční pocení. Zaznamenány byly tři závažné NÚ související s léčbou siltuximabem, a to infekce dolních cest dýchacích, anafylaktická reakce a sepse [4].
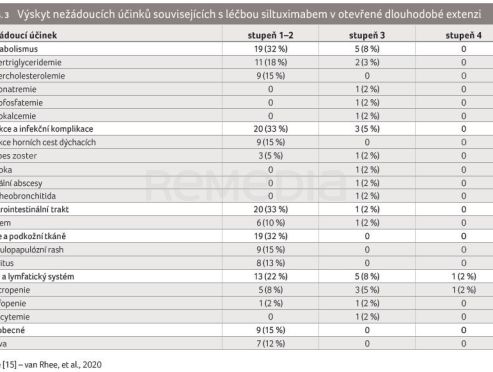 Dlouhodobá bezpečnost a snášenlivost siltuximabu byla hodnocena v otevřené dlouhodobé extenzi (LTE) studií fáze I a II, do které bylo zařazeno 60 pacientů (s výjimkou dvou nemocných s neresekabilní unicentrickou MCD měli všichni iMCD), u nichž nedošlo k progresi onemocnění během předchozích studií. Pacienti byli léčeni siltuximabem v dávce 11 mg/kg každé 3 týdny (aplikační interval mohl být prodloužen na 6 týdnů) [15]. V interim analýze LTE, která byla omezena na pacienty postoupivší ze studie fáze I (n = 19) a byla provedena po 2 letech od zahájení LTE, nebyla pozorována na dávce závislá ani kumulativní toxicita [16]. Finální analýza byla provedena při mediánu délky sledování 6 let (interkvartilové rozpětí [IQR] 5,11–7,76), kdy medián trvání léčby siltuximabem činil 5,5 let (IQR 4,26–7,14).
Dlouhodobá bezpečnost a snášenlivost siltuximabu byla hodnocena v otevřené dlouhodobé extenzi (LTE) studií fáze I a II, do které bylo zařazeno 60 pacientů (s výjimkou dvou nemocných s neresekabilní unicentrickou MCD měli všichni iMCD), u nichž nedošlo k progresi onemocnění během předchozích studií. Pacienti byli léčeni siltuximabem v dávce 11 mg/kg každé 3 týdny (aplikační interval mohl být prodloužen na 6 týdnů) [15]. V interim analýze LTE, která byla omezena na pacienty postoupivší ze studie fáze I (n = 19) a byla provedena po 2 letech od zahájení LTE, nebyla pozorována na dávce závislá ani kumulativní toxicita [16]. Finální analýza byla provedena při mediánu délky sledování 6 let (interkvartilové rozpětí [IQR] 5,11–7,76), kdy medián trvání léčby siltuximabem činil 5,5 let (IQR 4,26–7,14).
Celkem 58 % pacientů bylo léčeno siltuximabem v původní dávce 11 mg/kg každé 3 týdny, 42 % bylo převedeno na aplikaci každých 6 týdnů. Nejméně jeden NÚ byl hlášen u všech pacientů, přičemž souvislost s léčbou byla předpokládána v 88 % případů (tab. 3). Nejčastěji se jednalo o infekce horních cest dýchacích (52 %), průjem (38 %), nauzeu (37 %), rash (35 %) a artralgie (33 %). Nežádoucí účinky stupně ≥ 3 byly zaznamenány u 60 % pacientů, nejčastěji šlo o hypertenzi (13 %), únavu (8 %), nauzeu (7 %), neutropenii (7 %) a zvracení (5 %). Závažné NÚ se objevily u 25 (42 %) pacientů, pouze u dvou z nich však souvisely s léčbou siltuximabem (jednalo se o jeden případ polycythaemia vera a jeden případ retence moči). Při terapii siltuximabem byly pozorovány některé biochemické a hematologické laboratorní abnormality (zcela nejčastěji hypertriglyceridemie), většina z nich byla stupně 1–2. Kvůli NÚ ukončili léčbu siltuximabem pouze dva pacienti [15].
Protilátky proti siltuximabu byly detekovány celkem u tří (5 %) pacientů, u žádného z nich se však nejednalo o neutralizační protilátky. Přítomnost protilátek proti siltuximabu neměla vliv na jeho účinnost, bezpečnost ani farmakokinetiku [15].
Indikace a dávkování
Siltuximab je indikován k léčbě dospělých pacientů s multicentrickou formou Castlemanovy choroby a negativitou HIV a HHV‑8. Doporučená dávka je 11 mg/kg siltuximabu podávaná formou intravenózní infuze po dobu 1 hodiny jednou za 3 týdny až do selhání léčby. Během prvních 12 měsíců je zapotřebí před každou dávkou siltuximabu a následně každý třetí dávkovací cyklus provést hematologické laboratorní testy, a pokud nejsou splněna definovaná léčebná kritéria [11], je vhodné zvážit odložení léčby. Snižování dávky siltuximabu se nedoporučuje [11].
Závěr
Multicentrická Castlemanova choroba je vzácné onemocnění charakterizované generalizovanou lymfadenopatií a systémovými příznaky. U části pacientů vzniká MCD na podkladě infekce HHV‑8, až u dvou třetin pacientů však je etiopatogeneze onemocnění nejasná neboli idiopatická. V předchozích dvou desetiletích bylo toto onemocnění léčeno většinou léčebnými režimy pro B lymfomy, tedy kombinací rituximabu nejčastěji s alkylačními cytostatiky či doxorubicinem a glukokortikoidy; u mladých pacientek nebo pacientů jsme používali jen rituximab s kortikosteroidy bez cytostatik, aby nedošlo k poškození zárodečných buněk. Příchod léků ze skupiny IMID vedl k jejich testování také v indikaci CD a příznivým překvapením bylo zjištění, že tyto léky, dominantně používané pro léčbu mnohočetného myelomu, jsou účinné i v této indikaci.
V našem nevelkém souboru jsme pozorovali, že někteří nemocní reagovali na léky ze skupiny IMID lépe než na rituximab, zatímco u jiných pacientů bylo léčebné odpovědi dosaženo pomocí rituximabu při nedostatečné odpovědi na předchozí terapii léky ze skupiny IMID [17]. Tyto léky jsme používali takzvaně „off‑label“ neboli jen se schválením úhrady revizním lékařem. V letech 2020–2021 pak byla paleta léků určených pro Castlemanovu chorobu obohacena o první oficiální registrovaný lék – siltuximab.
U mnohých maligních chorob je možné prokázat určité mutace, například mutaci BRAF V600E u melanomu, ale také u histiocytózy z Langerhansových buněk či u Erdheimovy–Chesterovy choroby; průkaz mutace BRAF V600E signalizuje dobrou léčebnou odpověď na vemurafenib. U četných maligních chorob jsou tedy již známy prediktory léčebné odpovědi pro určité léky. V případě CD zatím nejsou známy žádné znaky, které by predikovaly léčebnou odpověď na určité léky. Nezbývá tedy nic jiného než vždy otestovat lék, o němž je známo, že jeho podání je spojeno s vysokým počtem léčebných odpovědí, a při jeho neúspěchu následně zkoušet další léky, o nichž je známo, že u této nemoci jsou účinné.
V současnosti tak mají lékaři na výběr zahájit léčbu siltuximabem nebo rituximabem v kombinaci s glukokortikoidy a případně s cytostatiky, a při nedostatečné léčebné účinnosti vyzkoušet léky ze skupiny IMID. Pokud ani léky ze skupiny IMID nevedou k léčebné odpovědi, je možné použít anakinru nebo další léky, jejichž přehled uvádíme v článku „Léčba multicentrické a unicentrické Castlemanovy choroby“ v časopisu Farmakoterapie 2021/3 [18]. Účinek podané léčby hodnotíme jak dle laboratorních ukazatelů, tak pomocí zobrazení FDG‑PET/CT [19].
Cílem tohoto článku je dominantně poukázat na významnou účinnost siltuximabu u nemocných s iMCD a jeho příznivý bezpečnostní profil, a to i při dlouhodobé aplikaci. Na základě výsledků citovaných studií byl siltuximab registrován pro terapii dospělých pacientů s multicentrickou formou Castlemanovy choroby a negativitou HIV a HHV‑8 a stal se jediným lékem schváleným v indikaci iMCD v Severní Americe a v Evropě.
Článek byl převzat se svolením autorů a časopisu z Farmakoterapie 2021; 17(3): 475–483. Publikujeme s drobnými formálními úpravami.
Literatura
[1] Brandt SJ, Bodine DM, Dunbar CE, et al. Dysregulated interleukin 6 expression produces a syndrome resembling Castleman’s disease in mice. J Clin Invest 1990; 86: 592–599.
[2] Yu L, Tu M, Cortes J, et al. Clinical and pathological characteristics of HIV‑ and HHV‑8‑negative Castleman disease. Blood 2017; 129: 1658–1668.
[3] Szturz P, Moulis M, Adam Z, et al. Castlemanova choroba. Klin Onkol 2011; 24: 424–434.
[4] van Rhee F, Wong RS, Munshi N, et al. Siltuximab for multicentric Castleman’s disease: a randomised, double‑blind, placebo controlled trial. Lancet Oncol 2014; 15: 966–974.
[5] Nishimoto N, Kanakura Y, Aozasa K, et al. Humanized anti‑interleukin‑6 receptor antibody treatment of multicentric Castleman disease. Blood 2005; 106: 2627–2632.
[6] Nemeth E, Rivera S, Gabayan V, et al. IL‑6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest 2004; 113: 1271–1276.
[7] Fajgenbaum DC, Uldrick TS, Bagg A, et al. International, evidence‑based consensus diagnostic criteria for HHV‑8‑negative/idiopathic multicentric Castleman disease. Blood 2017; 129: 1646–1657.
[8] Liu AY, Nabel CS, Finkelman BS, et al. Idiopathic multicentric Castleman’s disease: a systematic literature review. Lancet Haematol 2016; 3: e163–e175.
[9] van Rhee F, Voorhees P, Dispenzieri A, et al. International, evidence‑based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood 2018; 132: 2115–2124.
[10] Nishimoto N, Terao K, Mima T, et al. Mechanisms and pathologic significances in increase in serum interleukin‑6 (IL‑6) and soluble IL‑6 receptor after administration of an anti‑IL‑6 receptor antibody, tocilizumab, in patients with rheumatoid arthritis and Castleman disease. Blood 2008; 112: 3959–3964.
[11] Sylvant – Souhrn údajů o přípravku, 2019; https://www.sukl.cz.
[12] Kurzrock R, Voorhees PM, Casper C, et al. A phase I, open‑label study of siltuximab, an anti‑IL‑6 monoclonal antibody, in patients with B‑cell non‑Hodgkin lymphoma, multiple myeloma, or Castleman disease. Clin Cancer Res 2013; 19: 3659–3670.
[13] van Rhee F, Fayad L, Voorhees PM, et al. Siltuximab, a novel anti‑interleukin‑6 monoclonal antibody, for Castlemanʼs disease. J Clin Oncol 2010; 28: 3701–3708.
[14] van Rhee F, Rossi JF, Simpson D, et al. Newly diagnosed and previously treated multicentric Castleman disease respond equally to siltuximab. Br J Haematol 2021; 192: e28–e31.
[15] van Rhee F, Casper C, Voorhees PM, et al. Long‑term safety of siltuximab in patients with idiopathic multicentric Castleman disease: a prespecified, open‑label, extension analysis of two trials. Lancet Haematol 2020; 7: e209–e217.
[16] van Rhee F, Casper C, Voorhees PM, et al. A phase 2, open‑label, multicenter study of the long‑term safety of siltuximab (an antiinterleukin‑6 monoclonal antibody) in patients with multicentric Castleman disease. Oncotarget 2015; 6: 30408–30419.
[17] Adam Z, Szturz P, Krejčí M, et al. Léčba 14 případů Castlemanovy nemoci: zkušenosti jednoho centra a přehled literatury. Vnitř Lék 2016; 62: 287–298.
[18] Adam Z, Pour L, Štork M, et al. Léčba multicentrické a unicentrické Castlemanovy choroby. Farmakoterapie 2021; 17: 441–447.
[19] Koukalová R, Selingerová I, Řehák Z, et al. FDG‑PET/CT v diagnostice a hodnocení léčebné odpovědi Castlemanovy choroby – retrospektivní studie 29 případů z jednoho centra. Klin Onkol 2021; 34: 1–8.
Idiopatická Castlemanova choroba očima pacientky
Autentický příběh mladé pacientky je pro Castlemanovu chorobu typický. Karolína měla štěstí na neústupnou lékařku – revmatoložku –, která šla za objasněním známek zánětu (v tomto případě vysoké hodnoty CRP) až téměř ke stanovení diagnózy. Za obecným histopatologickým popisem lymfadenopatie nejasné etiologie se může skrývat léčitelná idiopatická Castlemanova choroba, jejíž patomechanismus je veden nadprodukcí prozánětlivého cytokinu – interleukinu 6 (IL‑6). Cesta k léčbě však nebyla jednoduchá. Kolik takových pacientů chodí mezi námi, aniž vědí, co jim je?
Jmenuji se Karolína a je mi 29 let. Narodila jsem se v Mostě. Po základní škole jsem šla na hotelovou střední školu a po úspěšném složení maturitní zkoušky jsem se přihlásila na Vysokou školu finanční a správní, kde jsem získala bakalářský titul. V roce 2015 jsem nastoupila do naší rodinné firmy jako administrativní pracovnice a expedientka zboží. Moje pracovní doba byla od 3:00 do 10:00 každý všední den, práce mě moc bavila a naplňovala a vlastně mě stále baví a naplňuje, jen se změnila má pracovní doba…
Úplně nenápadný začátek
Žila jsem běžným životem jako obyčejná holka: občas jsem si šla zacvičit, někdy zašla s přáteli na drink, v létě jezdila na inlinech atd. – stále takový život vedu, jen je tady jedno malé „ale“.
Bylo léto 2017 a já dostala angínu, která se projevovala klasickou bolestí v krku; předpokládala jsem, že dostanu antibiotika. Šla jsem ke svému obvodnímu lékaři, který mě prohlédl a řekl, že mám angínu a že mi vezme z prstu krev (CRP). Bylo to poprvé, kdy jsem se setkala s bodnutím do prstu. Hodnota CRP byla kolem 140, doktor se zalekl, odebral mi vzorek znova a neustále se vyptával, jestli mám teplotu, zimnici a nějaké další bolesti (kromě krku). Bylo mi dobře, teploty jsem neměla (zvýšenou teplotu na sobě poznám hned – většinou mám nižší než 36,2). Druhý vzorek opět ukázal hodnotu přes 100. Doktor mi dal antibiotika a šla jsem domů. Poté, co jsem antibiotika dobrala, jsem šla na kontrolu – hodnota CRP byla opět přes 100, ale já už neměla bolesti žádné, ani v krku. Přesto jsem opět dostala další antibiotika. Po dobrání druhých antibiotik, kdy moje střevní mikroflóra se už potřebovala dát trochu „dohromady“, jsem stále měla CRP přes 100. A tím začalo pátrání po tom, co vlastně tuto vysokou hodnotu způsobuje…
Případ vysokého CRP
Můj obvodní lékař skončil svou ordinaci a já jsem přešla k obvodní lékařce. Nejprve mi dělali neustále nějaké krevní rozbory: CRP stále 120–140, hemoglobin jsem měla nižší, možná kolem 100, a trombocyty naopak vyšší – až, pokud si dobře vzpomínám, 600. Nikdo netušil, co mi je. Bylo to pro mě psychicky velmi těžké období, kdy se člověk cítí zdravý, ale lékaři tvrdí pravý opak.
Poslali mě do nemocnice, kde jsem ležela dva dny na interně. Neustále mi brali krev a moč. Po dvou dnech mi pan primář sdělil, že se třeba časem něco objeví a že mohu domů. To mi psychicky nijak nepomohlo, ba naopak.
Obvodní lékařka mě poslala na revmatologii v Mostě a já jela ještě i do Prahy do Revmatologického ústavu. Prý může být příčina v kloubech. Ani v Mostě, ani v Praze však nic nenašli. Přesto se CRP paní doktorce‑revmatoložce nezdálo, prakticky se mě ujala a začala mě posílat na všelijaká vyšetření. Gynekologie, vyšetření prsou, ORL, zubní, kolonoskopie, gastroskopie, hematologie, vyšetření jícnu, sono břicha, CT kostí… I pak však nad mým vysokým CRP stále visel otazník. Zbývalo poslední vyšetření: PET/CT, které zobrazí celé tělo. Řekli mi: pokud se nic nenajde, necháme to být.
Strastiplná cesta k diagnóze
Nikdy nezapomenu na telefonát 19. ledna 2018. Byla jsem v práci a volá mi revmatoložka, že má výsledky a že se mnou chce mluvit. Okamžitě mi došlo, že se něco našlo, ale co? Chtěla se mnou mluvit osobně! Jen jsem seděla a brečela. V kanceláři byl můj tatínek a uklidňoval mě. Okamžitě se mnou jela moje maminka k paní revmatoložce Malecké (tímto bych jí chtěla za vše moc poděkovat). Oznámila mi, že se na PET/CT objevil nějaký útvar za hrudní kostí, ale že budu muset podstoupit biopsii toho útvaru.
S odstupem času zjišťuji, že plno těchto momentů mi jen pozvolna vystupuje z mlhy. Nedokážu slovy popsat své pocity, kdy se ze mě rázem stal nemocný člověk. Tolik strachu, pláče, bezmoci, probdělých nocí a samozřejmě vyhledávání na internetu, co mi asi je. Dnes vím, že na internetu se opravdu diagnózy nehledají, to nepřeji ani svému největšímu nepříteli.
Když mě v Ústí nad Labem přijímali na hrudní oddělení, paní doktorka mi oznámila, že se jedná o lymfom, že jen potřebují zjistit, o který typ jde, a že se to léčí chemoterapií.
Můj život, moje rodina, moje plány, sny a cíle, co teď bude dál? To se mi honilo hlavou a ještě teď mi tečou slzy, kdykoliv si na tento okamžik vzpomenu. Vidět svoji maminku, tatínka a sestru, jak nevědí, co mi říct, jak mě uklidnit a podpořit, že to bude dobré a že to zvládneme, když nikdo z nás netušil, co mě čeká a jak moc vážná moje diagnóza je.
Moje první narkóza – měla jsem strašný strach. Na krku mám malou jizvu a lidé si často myslí, že jsem po operaci štítné žlázy. Čekalo se na výsledky, bylo to nekonečné. Pak jednoho dne zazvonil telefon a já se dozvěděla, že z biopsie prakticky nic nevyšlo – jen lymfadenopatie. Ihned mi oznámili, že musím opět podstoupit operaci, tentokrát náročnější, kdy mi laparoskopicky vezmou kus uzliny mezi žebry. Vyfouknou plíci a pak ji zase nafouknou. Takže jsem zase na začátku. Kolotoč hysterického pláče a uklidňování se, to bylo na denním pořádku. Tohle období beru tak, že jsem vlastně nežila, jen se modlila, ať už to skončí a jsme zase ta veselá rodina jako předtím.
Když jsem se vzbudila po druhé narkóze, celý den se mi motala hlava, navíc jsem měla stále u sebe „kabelku“ (drén). Musela jsem dělat dechová cvičení. Moc jsem si přála jít domů, až mě po pár dnech pustili. Doma jsem se pak pár dní litovala, jak se rána hojila a měla jsem podrážděné nervy mezi žebry, bylo to dost bolestivé. Chyběla mi kapačka na bolest…
Diagnóza – konečně, ale…
Konečně jsem se dozvěděla výsledky – idiopatická forma Castlemanovy choroby. Já? Já jsem nemocná? Vždyť mi nic není. Nemocný člověk má přece bolesti a klinické příznaky, ale já ne. V Ústí mi doporučili lékaře v Praze nebo specialistu na toto onemocnění v Brně – pana profesora Adama. Ihned jsem se rozhodla pro Brno.
Moje první konzultace v Brně byla plná rozpaků. Najednou jsem se ocitla na hematoonkologické ambulanci. Už jen slovo „onko“ lidem nahání strach. Ale člověk to prostě musí přijmout: teď to tak je.
Pan profesor Adam na mě působil moc dobrým dojmem, profesionálním a lidským přístupem. Myslím si, že pro pacienta je strašně důležité důvěřovat svému lékaři. Vše mi vysvětlil, co vlastně ta moje choroba je, že není jasné, proč u mě propukla nebo zda ji mám od narození. Doteď pan profesor nechápe, jak jsem mohla s tak vysokým CRP fungovat.
Domluvili jsme se na biologické léčbě rituximabem. Zažádali jsme pojišťovnu o schválení, zamítnuto – prý mám dostat chemoterapii. Odmítala jsem ji, můj zdravotní stav nebyl tak vážný a bylo mi 25 let, chtěla jsem do budoucna rodinu.
S právníkem jsme se odvolali a poté mi byla léčba konečně schválena, tento proces určitě trval 3–4 měsíce.
… konečně léčba!
V květnu 2018 jsem byla hospitalizována ve FN Brno‑Bohunice, kdy mi poprvé podali rituximab pod dohledem – kvůli riziku alergické reakce. Vše proběhlo bez komplikací a já jezdila každých 14 dní na další dávku. Celkem to bylo osmkrát. Poslední infuzi jsem dostala v srpnu 2018. Výsledky byly dobré, hodnoty se srovnaly. Následující čtyři roky jsem jezdila na kontroly, léčbu jsem neměla. Ale časem začalo CRP opět růst.
 Přestože jsem měla CRP a další laboratorní testy opět vyšší nebo naopak nižší, než by měly být, cítila jsem se dobře. Ale pan profesor mi doporučil zase nasadit léčbu, tentokrát siltuximab, který v České republice ještě nebyl aplikován při idiopatické formě Castlemanovy choroby. Opět jsme zažádali pojišťovnu o schválení, ale opět byla léčba zamítnuta s tím, že mám dostat rituximab. Pan profesor Adam nelenil a kontaktoval distributora léku siltuximab s tím, že mi jej pojišťovna zamítla uhradit.
Přestože jsem měla CRP a další laboratorní testy opět vyšší nebo naopak nižší, než by měly být, cítila jsem se dobře. Ale pan profesor mi doporučil zase nasadit léčbu, tentokrát siltuximab, který v České republice ještě nebyl aplikován při idiopatické formě Castlemanovy choroby. Opět jsme zažádali pojišťovnu o schválení, ale opět byla léčba zamítnuta s tím, že mám dostat rituximab. Pan profesor Adam nelenil a kontaktoval distributora léku siltuximab s tím, že mi jej pojišťovna zamítla uhradit.
Netušila jsem, že zázraky se dějí, ale mně se jeden opravdu stal – dostala jsem možnost aplikování siltuximabu na svoji nemoc. Jsem za to vděčná a moc děkuji.
V květnu 2022 jsem byla na první aplikaci opět hospitalizována ve FN Brno‑Bohunice, s otazníkem, jak na mě látka bude působit. Žádné nežádoucí účinky, po první dávce CRP neuvěřitelně kleslo na hodnotu 2. I ostatní parametry se začaly zlepšovat a v současnosti jsou v normě. Prostě zázrak.
Nyní jezdím každé tři týdny na aplikaci – je ambulantní a trvá hodinu až dvě.
Cena běžného života
Castlemanova choroba mi hodně vzala – nejvíc asi čas, kdy jsem si stále pokládala otázky: Proč já? Proč jsem nemocná? Vzala mi na pár měsíců úsměv a radost, kdy jsem žila ve strachu, a také pár litrů krve při nekonečných odběrech, ale také mi hodně dala, třeba že zdraví není samozřejmost a že je možná občas dobré zajít na vyšetření nebo lékařskou prohlídku, že se dá žít plnohodnotný život i s nemocí, že dostávám lék, který do budoucna pomůže dalším nemocným lidem, setkala jsem se s úžasným lékařem – panem profesorem Adamem – a také se konečně podívala do Brna…
Ale hlavně žiju zase běžný život, chodím každý den do práce, jen ne ve tři ráno, ale v půl páté. Chodím si zacvičit, ráda jezdím na výlety a výšlapy, trávím čas se svým báječným přítelem Romanem, který je mi oporou a parťákem do života a s nímž plánuji rodinu. A už jsme zase ta veselá rodina, kdy moji nejbližší vidí, že jsem v pořádku, zdravá a šťastná. Cítím však, jak moc důležitá pro mne byla podpora celé rodiny, která se mnou absolvovala celou mou cestu k normálnímu životu. Všem jim patří veliký dík.
S láskou Karolína
Medicína po promoci 4/2022
Kazuistika pacienta s Castlemanovým syndromem z Klinického centra Univerzity v Debrecínu
Maďarské vydavatelství Medical Tribune uveřejnilo kazuistiku autorů Boglárky Brúgós a György Pflieglera o pacientovi s Castlemanovou chorobou (CD), který je léčen od roku 2016 inhibitorem interleukinu 6 siltuximabem. Do roku 2020 trvala po úvodní sérii léčby siltuximabem remise, relaps se podařilo opět zvládnout sérií infuzí se siltuximabem. Text maďarských autorů uveřejňujeme v překladu původního textu s jejich laskavým svolením a svolením vydavatelství Medical Tribune v Maďarsku.
Úvod
Castlemanova choroba je vzácné heterogenní lymfoproliferativní onemocnění se známými unicentrickými (UCD) a multicentrickými formami (MCD) [1]. Pro UCD je charakteristické izolované zvětšení lymfatických uzlin, které je často asymptomatické, ale mohou se objevit lokální příznaky. Avšak u MCD jsou časté systémové příznaky, jako jsou například klasické, tzv. B symptomy (horečka, úbytek hmotnosti, noční pocení), generalizovaná lymfadeno‑, hepato‑ a splenomegalie [2]. Prostřednictvím své kazuistiky chtějí autoři poukázat na některé důležité aspekty diagnostiky a léčby CD.
Kazuistika
Třiašedesátiletý pacient má v anamnéze diabetes mellitus 1. typu a hypertenzi od roku 2007. V červnu 2015 bylo zahájeno vyšetřování pro výrazný úbytek hmotnosti, normocytární anémii vyžadující transfuzi a pro zvýšenou sedimentaci erytrocytů (112 mm/h). Karence jako příčina anémie nebyla potvrzena, Coombsův test byl negativní. Ultrazvuk břicha a vyšetření počítačovou tomografií (CT) odhalily středně závažnou hepatosplenomegalii, retroperitoneální lymfadenomegalii a infiltraci pankreatu. Byla popsána destrukce dorzální části těla sedmého krčního obratle s epidurální propagací. Rentgenový snímek hrudníku byl negativní. Fyzikální vyšetření odhalilo krční lymfadenomegalii. Při podrobném vyšetření byly při gastroskopii zjištěny drobné eroze v antru žaludku, kolonoskopie byla z technických důvodů úspěšná jen částečně, ale malignita ve vyšetřeném úseku nebyla nalezena. Ultrazvukové vyšetření srdce prokázalo malý perikardiální výpotek. Scintigrafie kostí neprokázala lytické léze, v elektroforéze séra nebyla nalezena monoklonální gamapatie a byl vyloučen imunodeficit. Urologické vyšetření potvrdilo benigní hyperplazii prostaty. Onemocnění štítné žlázy bylo vyloučeno a systémové autoimunitní onemocnění se jevilo také jako nepravděpodobné kvůli chybějícím klinickým symptomům a negativitě imunosérologických vyšetření. V kostní dřeni nebyla nalezena známka malignity. Virologické vyšetření prokázalo séropozitivitu pro toxoplazmózu a virus hepatitidy C.
Koncem listopadu 2015 byl odebrána biopsie jedné z krčních lymfatických uzlin a histologickým vyšetřením byla stanovena diagnóza CD hyalinně vaskulárního typu. Polymerázová řetězová reakce (PCR) ukázala negativitu lidského herpesviru 8 (HHV‑8).
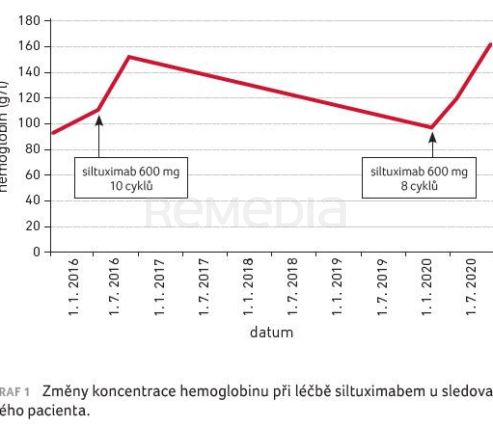 Výše uvedená diagnóza byla potvrzena v květnu 2016 a v červnu 2016 byl pacient přijat na oddělení vzácných onemocnění, kde byl podáván vinblastin v dávce 4 mg/m2 (4 cykly každé 2 týdny) a metylprednisolon v dávce 1 mg/kg. S ohledem na patomechanismus onemocnění byla předložena žádost o individuální úhradu léčby inhibitorem interleukinu 6 (IL‑6) – siltuximabem. Koncentrace IL‑6 nebyla z technických důvodů stanovena. Vzhledem k anti‑HCV (virus hepatitidy C) pozitivitě bylo také zahájeno podávání antivirotika entekaviru v dávce 0,5 mg denně. Po schválení úhrady dostává pacient první cyklus siltuximabu (600 mg) v září 2016. V období od září 2016 do června 2017 dostal 10 infuzí siltuximabu, poté byl asymptomatický, tento stav trval až do března 2020. Krevní obraz byl stabilizován (graf 1).
Výše uvedená diagnóza byla potvrzena v květnu 2016 a v červnu 2016 byl pacient přijat na oddělení vzácných onemocnění, kde byl podáván vinblastin v dávce 4 mg/m2 (4 cykly každé 2 týdny) a metylprednisolon v dávce 1 mg/kg. S ohledem na patomechanismus onemocnění byla předložena žádost o individuální úhradu léčby inhibitorem interleukinu 6 (IL‑6) – siltuximabem. Koncentrace IL‑6 nebyla z technických důvodů stanovena. Vzhledem k anti‑HCV (virus hepatitidy C) pozitivitě bylo také zahájeno podávání antivirotika entekaviru v dávce 0,5 mg denně. Po schválení úhrady dostává pacient první cyklus siltuximabu (600 mg) v září 2016. V období od září 2016 do června 2017 dostal 10 infuzí siltuximabu, poté byl asymptomatický, tento stav trval až do března 2020. Krevní obraz byl stabilizován (graf 1).
V březnu 2020 se opět objevuje anémie, což vzbuzuje podezření na relaps aktivní CD. CT hrudníku bylo negativní, ale CT břicha odhalilo hepatosplenomegalii a lymfadenomegalii. Po schválení úhrady byla léčba siltuximabem znovu zahájena a pacient dostává osm cyklů, poslední v listopadu 2020. Od té doby je asymptomatický.
Diskuse
Castlemanova choroba je vzácné heterogenní lymfoproliferativní onemocnění se známou unicentrickou (UCD) a multicentrickou (MCD) formou, která se dále dělí do tří skupin podle toho, zda současně ne/existují další příznaky nebo jasný patologický faktor: idiopatická forma, HHV‑8 asociované onemocnění a POEMS (polyneuropatie, organomegalie, endokrinopatie, monoklonální gamapatie, kožní symptomy) asociované s MCD [1,3]. Nejčastějšími laboratorními abnormalitami jsou nespecifická anémie, trombocytopenie, hypoalbuminemie, hypergamaglobulinemie, zvýšená koncentrace proteinů akutní fáze a IL‑6. Pro diagnostiku je důležité histologické vyšetření a vyloučení diagnózy lymfomu.
Na základě histologického vzhledu se rozlišují tři typy: hyalinně (hyper) vaskulární, plazmocelulární a smíšený typ [1]. Nejčastěji se u UCD vyskytuje hyalinně vaskulární typ, v lymfatických uzlinách je pozorována kapsulární fibróza a kapilární proliferace pronikající do atrofických zárodečných center, lymfocyty jsou často lokalizovány „cibulovitě“ kolem dendritických buněk [2–4]. Tento typ se vyskytuje také u idiopatických MCD. U UCD se doporučuje primárně kurativní regionální resekce, ale v některých případech je opodstatněná i systémová léčba.
Velkým pokrokem v léčbě MCD je použití inhibitorů IL‑6 v první linii (siltuximab, tocilizumab) [1,2,4]. Alternativně lze použít anti‑CD20 protilátku – rituximab – nebo chemoterapii. O prognóze onemocnění je k dispozici jen málo údajů. Pětileté přežití pacientů s idiopatickou MCD je 100 % [5].
Histologické vyšetření sledovaného pacienta prokázalo hyalinně vaskulární typ, multicentrická forma byla potvrzena rozsáhlou lymfadenomegalií, systémovými příznaky a postižením dalších orgánů. Asociace s HHV‑8 byla vyloučena. Podle současných doporučení byla zahájena systémová kortikoterapie doplněná anti‑IL‑6 monoklonální protilátkou – siltuximabem (11 mg/kg každé 3 týdny i.v.). Léčba byla dobře tolerována a převládaly známé nežádoucí účinky jako mírná trombocytopenie a hyperlipidemie. Pacient dobře reagoval na léčbu a jeho systémové příznaky ustoupily. K relapsu došlo po pěti letech remise, ale po opakované léčbě lékem první volby nastala opět remise.
Castlemanova choroba je vzácné nezhoubné lymfoproliferativní onemocnění. Je‑li co nejdříve vysloveno podezření na CD a následně potvrzena diagnóza, lze u CD vhodnou moderní léčbou navodit úplnou remisi a pětileté přežití činí až 100 %.
Dr. Boglárka Brúgós, PhD; Dr. György Pfliegler
Expertní centrum pro vzácná onemocnění Univerzity v Debrecíně
Ústav interního lékařství, Oddělení vzácných onemocnění
Literatura
[1] Dispenzieri A, Fajgenbaum DC. Overview of Castleman disease. Blood 2020; 135: 1353−1364.
[2] Chan K‑L, Lade S, Prince HM, et al. Update and new approaches in the treatment of Castleman disease. J Blood Medicine 2016; 7: 145−158.
[3] Pfliegler György, Brúgós Boglárka. A Castleman‑betegség és terápiás lehetőségei. Medical Tribune 2021; 19: 22−25.
[4] Pribyl K, Vakayil V, Farooqi N, et al. Castleman disease: A single‑center case series. Int J of Surgery Case Reports 2021; 80: 1−7.
[5] Oksenhendler E, Boutboul D, Fajgenbaum D, et al. The full spectrum of Castleman disease: 273 patients studied over 20 years. Br J Haematol 2018; 180: 206−216.
Text byl uveřejněn s laskavým svolením autorů a vydavatelství Medical Tribune HU. Vyšlo v Medical Tribune 18. 1. 2022.
Státní ústav pro kontrolu léčiv rozhodl o úhradě léčby siltuximabem (Sylvant) v indikaci multicentrické Castlemanovy choroby
SÚKL stanovil přípravku v silách 100 mg a 400 mg úhradu ze zdravotního pojištění a zařadil jej do skupiny v zásadě terapeuticky zaměnitelných léčivých přípravků s obsahem léčivé látky siltuximab pro léčbu pacientů se symptomatickou idiopatickou multicentrickou Castlemanovou chorobou. Siltuximab je hrazen v terapii dospělých pacientů s multicentrickou formou Castlemanovy choroby, kteří jsou negativní na virus lidské imunodeficience (HIV) a lidský herpesvirus 8 (HHV‑8). Léčba je hrazena do selhání léčby nebo do nepřijatelné toxicity.
Preskripce s úhradou je omezena na specializovaná pracoviště.
Zdroj: SÚKL č. j. sukl216902/2024 ze dne 28. 8. 2024; https://www.sukl.cz